Metilmalonik asidemi - Methylmalonic acidemia
| Metilmalonik asidemi | |
|---|---|
| Diğer isimler | MMA |
 | |
| Metilmalonik asit | |
| Uzmanlık | Endokrinoloji |
Metilmalonik asidemi, olarak da adlandırılır metilmalonik asidüri,[yardım 1] bir otozomal çekinik[1] metabolik bozukluk normal amino asit metabolizmasını bozan.[2] Klasik bir tür organik asidemi.[3] Bu durumun sonucu, belirli yağların ve proteinlerin düzgün bir şekilde sindirilememesidir ve bu da toksik bir seviyede birikmesine yol açar. metilmalonik asit Kanın içinde.[4]
Metilmalonik asidemi, çeşitli genotipler,[5] genellikle erken teşhis edilen bozukluğun tüm biçimleri yenidoğan dönem, ilerleyen sunum ensefalopati ve ikincil hiperamonyemi. Hastalık teşhis edilmezse veya tedavi edilmezse ölümle sonuçlanabilir. Bu bozukluğun 48.000 doğumda 1 sıklığı olduğu tahmin edilmekle birlikte, teşhis edilen vakalardaki yüksek ölüm oranı kesin tespiti zorlaştırmaktadır.[4] Metilmalonik asidemiler, etnik sınırlar arasında eşit sıklıkta bulunur.[6]
Belirtiler ve işaretler
Etkilenen gen (ler) e bağlı olarak, bu bozukluk hafif ila yaşamı tehdit eden semptomlar sunabilir.
- İnme[4]
- Progresif ensefalopati[4]
- Nöbet[4][7]
- Böbrek yetmezliği[4][8]
- Kusma[4][7][8]
- Dehidrasyon[4][7][8]
- Gelişememe ve gelişimsel gecikmeler[4][7][8]
- Letarji[4][7][8]
- Tekrarlanan Maya enfeksiyonları[4]
- Asidoz[7]
- Hepatomegali[7][8]
- Hipotoni[7][8]
- Pankreatit[8]
- Solunum zorluğu[7]
Sebep olmak
Genetik

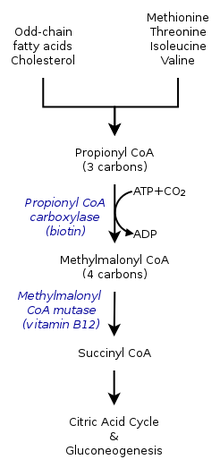
Metilmalonik asideminin kalıtsal formları, metabolik yol burada metilmalonil-koenzim A (CoA), metilmalonil-CoA mutaz enzimi tarafından süksinil-CoA'ya dönüştürülür.[9]
B vitamini12 metilmalonil-CoA'nın Süksinil-CoA'ya dönüşümü için de gereklidir. B vitamini kusurlarına yol açan mutasyonlar12 metabolizma veya taşınmasında sıklıkla metilmalonik asidemi gelişmesine neden olur.
Bu bozukluğun otozomal resesif kalıtım modeli vardır, bu da kusurlu genin bir otozom ve genin iki kopyası - her bir ebeveynden bir tane - hastalıktan etkilenmesi için kalıtsal olmalıdır. Otozomal resesif bozukluğu olan bir çocuğun ebeveynleri, kusurlu genin bir kopyasının taşıyıcılarıdır, ancak genellikle hastalıktan etkilenmezler.
Beslenme
Her zaman kalıtsal versiyonlarla birlikte gruplandırılmasa da, ciddi bir B vitamini eksikliği12 ayrıca genetik metilmalonik asidemilerle aynı semptom ve tedavilere sahip sendromla sonuçlanabilir.[10] Metilmalonil CoA, B vitamini gerektirir12 süksinil-CoA oluşturmak için. B miktarı12 kofaktör metilmalonil-CoA'nın süksinil-CoA'ya dönüşümü için yetersizdir, kullanılmayan metilmalonil-CoA oluşumu sonunda metilmalonik asidemiye yol açar. Bu tanı genellikle B vitamini göstergesi olarak kullanılır.12 eksiklik serum.[11]
Mekanizma
Patofizyoloji
Metilmalonik asidemide, vücut, amino asitler metiyonin, treonin, izolösin ve valin; sonuç olarak metilmalonik asit kanda ve dokularda birikir. Bu rahatsızlıktan muzdarip olanlar ya işlevsel kopyalardan yoksundur ya da aşağıdaki enzimlerin bir ya da daha fazlasının yeterli seviyelerinde: metilmalonil CoA mutaz, metilmalonil CoA epimeraz veya dahil olanlar adenosilkobalamin sentez.[7][8]
Metilmalonil CoA mutaz
Vakaların% 60 kadarının mutasyona uğramış bir sonuç olduğu tahmin edilmektedir. MUT protein metilmalonil CoA mutazını kodlayan gen. Bu enzim, öncelikle yukarıda bahsedilen amino asitlerin ve yağların parçalanmasının potansiyel olarak toksik türevlerinin sindirilmesinden sorumludur. kolesterol,[8] özellikle bu enzim dönüştürür metilmalonil-CoA içine süksinil-CoA.[12] Bu enzim olmadan vücudun metilmalonik asidi ve ilgili bileşikleri nötralize etme veya uzaklaştırma yolu yoktur. Bu enzimin etkisi aynı zamanda içindeki mutasyonlar tarafından da sakatlanabilir. MMAA, MMAB, ve MMADHC her biri metilmalonil CoA mutazının normal işleyişi için gerekli bir proteini kodlayan genler.[8]
Metilmalonil CoA epimeraz
Mutasyonlar MCEE Metilmalonil CoA epimeraz proteinini kodlayan, aynı zamanda metilmalonil rasemaz olarak da anılan gen, bozukluğun ilgili metalonil CoA mutaz varyantından çok daha hafif bir formuna neden olacaktır. Mutaz gibi, epimeraz da aynı maddeleri parçalama işlevini görür, ancak mutazın yaptığından önemli ölçüde daha az ölçüde.[8] fenotipik Mutaza karşı epimeraz eksikliğinden kaynaklanan farklılıklar o kadar hafiftir ki, bu genetik eksikliğin bir bozukluk veya klinik sendrom olarak kabul edilip edilemeyeceği konusunda tıp camiasında tartışma vardır.[13]
Adenosilkobalamin
B vitamini olarak da bilinir12, bu kobalamin formu gereklidir kofaktör metilmalonil CoA mutaz. Fizyolojik olarak normal seviyelerde enzimin fonksiyonel bir versiyonunda bile, eğer B12 bu aktif forma dönüştürülemezse, mutaz işlev göremez.[8]
İlerleme
Hastalığın farklı aşamaları olmamasına rağmen, Metilmalonik asidemi ilerleyici bir durumdur; Bu bozukluğun semptomları, metilmalonik asit konsantrasyonu arttıkça artar. Tetikleyici proteinler ve yağlar diyetten çıkarılmazsa, bu birikim onarılamaz böbrek veya karaciğer hasarına ve sonunda ölüme yol açabilir.[4]
Teşhis
Organik asideminin en yaygın formlarından biri değilse de,[14] Metilmalonik asidemi, bebeğin diyetine proteinler eklenene kadar semptomlar genellikle kendiliğinden ortaya çıkmadığı için doğumda belirgin değildir.[4] Bu nedenle semptomlar tipik olarak yaşamın ilk yılı içinde herhangi bir zamanda ortaya çıkar.[14] Tanı konulmadığında bu bozukluğun komplikasyonlara neden olabileceği ciddiyet ve hız nedeniyle, metilmalonik asidemi taraması genellikle yenidoğan tarama muayenesine dahil edilir.[4][15]
Amino asitleri tam olarak parçalayamama nedeniyle, protein sindiriminin yan ürünü olan metilmalonik asit bileşiği, etkilenenlerin kanında ve idrarında orantısız bir konsantrasyonda bulunur. Bu anormal seviyeler, bozukluğun teşhisi için ana tanı kriterleri olarak kullanılır. Bu bozukluk tipik olarak bir idrar analizi veya kan paneli.[14] Metilmalonik asideminin varlığından da şüphelenilebilir. CT veya MRI taraması veya amonyak testi, ancak bu testler hiçbir şekilde spesifik değildir ve klinik ve metabolik / korelasyon gerektirir.[4] Yüksek seviyeleri amonyak, glisin, ve keton cisimleri kan ve idrarda da mevcut olabilir.[7]
Türler
Metilmalonik asidemi, bozukluğun kalıtsal formuna neden olan spesifik genetik mutasyon tarafından belirlenen çeşitli teşhisler, tedavi gereksinimleri ve prognozlara sahiptir.[5] Aşağıdakiler metilmalonik asidemiden sorumlu bilinen genotiplerdir:
| OMIM | İsim | Gen |
|---|---|---|
| 251100 | cblA türü | MMAA |
| 251110 | cblB türü | MMAB |
| 277400 | cblC türü | MMACHC |
| 277410 | cblD türü | MMADHC[16] |
| 277380 | cblF türü | LMBRD1[17] |
| 251000 | mut türü | MUT |
Mut tipi ayrıca, mutO ve mut-alt tiplerine bölünebilir; mutO, metilmalonil CoA mutazının tam eksikliği ve daha şiddetli semptomlarla karakterize edilir ve mutaz aktivitesinin azalması ile karakterize edilir.[6]
Metilmalonik asideminin Mut-, cblB ve cblA versiyonlarının kobalamin duyarlı olduğu bulunmuştur. Mut0, yanıt vermeyen bir değişkendir.[6]
Tedavi
Diyet
Bu durumun tüm formları için tedavi, öncelikle düşük proteinli bir diyete ve bireyin muzdarip olduğu bozukluğun varyantına bağlı olarak çeşitli diyet takviyelerine dayanır. Tüm varyantlar yanıt verir levo izomeri nın-nin karnitin Etkilenen maddelerin uygun olmayan şekilde parçalanması, hastaların karnitin eksikliği geliştirmesine neden olur. Karnitin ayrıca, düşük proteinli diyetlerde birikmesi yaygın olan açil-CoA'nın idrarla atılabilen açil-karnitine dönüştürülerek uzaklaştırılmasına da yardımcı olur. Metilmalonil asideminin tüm biçimleri kobalamine yanıt vermemesine rağmen, siyanokobalamin takviyeleri genellikle bu bozukluğun birinci basamak tedavisinde kullanılır.[12] Birey hem kobalamin hem de karnitin takviyelerine yanıt verirse, o zaman Mayıs küçük miktarlarda sorunlu amino asit izolösin, treonin, metiyonin ve valin içeren maddeleri bir saldırıya neden olmadan almaları mümkün olabilir.[4]
Cerrahi
Daha aşırı bir tedavi, durumu olmayan bir donörden böbrek veya karaciğer naklini içerir. Yabancı organlar, kusurlu enzimlerin işlevsel bir versiyonunu üretecek ve metilmalonik asidi sindirecektir, ancak organ naklinin tüm dezavantajları bu durumda da uygulanabilir.[4] Merkezi sinir sisteminin vücudun geri kalanından izole edilmiş bir sistemde metilmalonik-CoA'yı metabolize edebileceğini gösteren kanıtlar vardır. Durum böyleyse, transplantasyon metilmalonik asidin transplantasyondan önceki nörolojik etkilerini tersine çeviremeyebilir veya sürekli birikimle beyinde daha fazla hasarı önleyemez.[18][12]
Prognoz
Prognoz, durumun ciddiyetine ve kişinin tedaviye verdiği yanıta bağlı olarak değişecektir. Prognoz, kobalamine duyarlı varyantlara sahip olanlar için tipik olarak daha iyidir ve kobalamine duyarlı olmayan varyantlardan muzdarip olanlarda umut vermez,[12] tipik olarak daha hafif varyantlar, daha şiddetli varyantlara göre popülasyonda daha yüksek bir sıklığa sahiptir.[14] Diyet değişikliği ve sürekli tıbbi bakımla bile, yanıt vermeyen asidemi olanlarda nörolojik hasarı önlemek mümkün olmayabilir.[12] Uygun tedavi veya teşhis olmaksızın, ilk asidemik atağın ölümcül olması nadir değildir.[4]
Bu zorluklara rağmen, ilk olarak 1967'de tanımlandığından bu yana, durumun tedavisi ve anlaşılması, yanıt vermeyen metilmalonik asidemi formlarına sahip olanların bile yetişkinliğe ulaşabilmeleri ve hatta çocukları taşıyıp doğurabilmeleri için duyulmamış bir noktaya geldi. güvenli bir şekilde.[18]
Araştırma
Nozolojik tarih
MMA ilk olarak Oberholzer ve ark.[19] 1967'de.[18]
Nörolojik etkiler
MMA'nın sinir sistemi üzerinde feci etkileri olabileceği uzun süredir bildirilmiştir; ancak bunun meydana geldiği mekanizma hiçbir zaman belirlenmemiştir. 15 Haziran 2015'te yayınlanan araştırma, metilmalonik asidin, benzer bir alternatif asitle tedavi edilen bir kontrol grubu nöron kullanılarak in vitro ortamda fetal sıçanlardan izole edilen nöronlar üzerindeki etkileri üzerine gerçekleştirildi. pH. Bu testler, metilmalonik asidin hücresel boyutta azalmaya ve hücresel hızda artışa neden olduğunu göstermiştir. apoptoz konsantrasyona bağlı bir şekilde, daha yüksek konsantrasyonlarda daha aşırı etkiler görülür. Ayrıca, bu tedavi edilen nöronların mikro dizi analizi de bunu bir epigenetik -düzey metilmalonik asit, özellikle apoptozla ilgili olanlar dahil olmak üzere 564 genin transkripsiyon oranını değiştirir, s53 ve MAPK sinyalleme yolları.[20]
Mitokondriyal disfonksiyon
Metilmalonil-CoA'nın süksinil-CoA'ya dönüşümü, mitokondri azalmış bir sonucu olarak mitokondriyal disfonksiyon elektron taşıma zinciri işlevinden uzun süredir MMA'da bir özellik olduğundan şüpheleniliyor. Son[ne zaman? ] Araştırmalar, sıçan modellerinde hastalıktan etkilenen sıçanların mitokondrilerinin megamitokondri olarak adlandırılan olağandışı boyuta ulaştığını buldu. Bu megamitokondriler ayrıca deforme iç yapılara ve iç yapılarında elektron zenginliğinde bir kayba sahip gibi görünmektedir. matris. Bu megamitokondriler ayrıca, özellikle sadece yaklaşık% 50 verimlilikte işlev gören solunum kompleksi IV'te solunum zinciri fonksiyonunda azalma belirtileri gösterdi. MMA'dan muzdarip 5 yaşındaki bir çocuktan transplant sırasında alınan bir karaciğer örneğinin mitokondrilerinde de benzer değişiklikler tespit edildi.[21]
İyi huylu mut fenotip
Son[ne zaman? ] p.P86L olarak adlandırılan spesifik bir mutasyona sahip yanıt vermeyen mutO MMA sunan birkaç hastadaki vaka çalışmaları, mut tip MMA'da daha fazla alt bölüm olasılığının mevcut olabileceğini düşündürmektedir. Bunun spesifik mutasyondan mı yoksa erken teşhis ve tedaviden mi kaynaklandığı şu anda belirsiz olsa da, kobalamin takviyelerine tam yanıt verilmemesine rağmen, bu bireylerin büyük ölçüde iyi huylu ve neredeyse tamamen asemptomatik bir MMA versiyonu geliştirdiği görülmüştür. Sürekli olarak kanda ve idrarda yüksek metilmalonik asit göstermesine rağmen, bu bireylerin büyük bir kısmı gelişimsel olarak normal göründü.[22]
Önemli durumlar
- St.Louis bebeği olan Ryan Stallings'e yanlışlıkla etilen glikol zehirlenmesi 1989'da MMA yerine, yanlış bir cinayet mahkumiyeti ve annesi için ömür boyu hapis cezasına yol açan, Patricia Stallings.[18]
Ayrıca bakınız
Notlar
- ^ İsimler metilmalonik asidemi ve metilmalonik asidüribazen katı olarak da yazılan Bileşikler (metilmalonikasidemi ve metilmalonikasidüri), son ekleri kullanın -emia ve -üri ve kelimenin tam anlamıyla "[fazla] anlamına gelir metilmalonik asit içinde kan "ve" [fazlalık] metilmalonik asit içinde idrar "sırasıyla; hem sıvı analizi bulgularını hem de bunlara neden olan hastalık varlığını etiketlemek için kullanılırlar.
Referanslar
- ^ Radmanesh, A; Zaman, T; Ganaati, H; Molaei, S; Robertson, Rl; Zamani, Aa (Temmuz 2008). "Metilmalonik asidemi: 52 çocukta beyin görüntüleme bulguları ve literatürün gözden geçirilmesi". Pediatrik Radyoloji. 38 (10): 1054–61. doi:10.1007 / s00247-008-0940-8. PMID 18636250. S2CID 24915585.
- ^ "MMA Çalışması: Çalışmamız Hakkında SSS". genome.gov. Alındı 26 Nisan 2016.
- ^ Dionisi-Vici C, Deodato F, Raschinger W, Rhead W, Wilcken B (2006). "Klasik organik asidüri, propiyonik asidüri, metilmalonik asidüri ve izovalerik asidüri: tandem kütle spektrometresi kullanılarak genişletilmiş yenidoğan taramasının uzun vadeli sonuçları ve etkileri". J Inherit Metab Dis. 29 (2–3): 383–389. doi:10.1007 / s10545-006-0278-z. PMID 16763906. S2CID 19710669.
- ^ a b c d e f g h ben j k l m n Ö p q r "Metilmalonik asidemi: MedlinePlus Tıbbi Ansiklopedisi". www.nlm.nih.gov. Alındı 2015-10-27.
- ^ a b Matsui, Sm; Mahoney, Mj; Rosenberg, Le (Nisan 1983). "Kalıtsal metilmalonik asidemilerin doğal geçmişi" (Ücretsiz tam metin). New England Tıp Dergisi. 308 (15): 857–61. doi:10.1056 / NEJM198304143081501. ISSN 0028-4793. PMID 6132336.
- ^ a b c "MMA Çalışması: Genel Bilgiler". www.genome.gov. Alındı 2015-11-03.
- ^ a b c d e f g h ben j k "Asidemi, Metilmalonik - NORD (Nadir Bozukluklar Ulusal Örgütü)". NORD (Ulusal Nadir Bozukluklar Örgütü). Alındı 2015-10-29.
- ^ a b c d e f g h ben j k l m "Metilmalonik asidemi". Genetik Ana Referans. 2015-10-26. Alındı 2015-11-02.
- ^ Sakomoto O, Ohura T, Matsubara Y, Takayanagi M, Tsuchiya S (2007). "Metilmalonik asidemili Japon hastalarda MUT geninin mutasyon ve haplotip analizleri". İnsan Genetiği Dergisi. 52 (1): 48–55. doi:10.1007 / s10038-006-0077-2. PMID 17075691.
- ^ Higginbottom MC, Sweetman L, Nyhan WL (1978). "Metilmalonik asidüri sendromu, homosistinüri, megaloblastik anemi ve B vitamininde nörolojik anormallikler12-sıkı bir vejeteryanın yetersiz emzirilen bebeği ". N Engl J Med. 299 (7): 317–323. doi:10.1056 / NEJM197808172990701. PMID 683264.
- ^ http://www.biology.arizona.edu/biochemistry/problem_sets/b12/04t.html
B vitamini12 eksikliği - Metilmalonik asidüri bağlantısı - ^ a b c d e "Metilmalonik Asidemi: Metilmalonik Asidemi, Etiyoloji ve Nöropatolojiye Kısa Bir Bakış, Metilmalonik Asideminin Değerlendirilmesi". 2019-03-05. Alıntı dergisi gerektirir
| günlük =(Yardım) - ^ "OMIM Giriş - # 251120 - METİLMALONİL-CoA EPIMERAZ EKSİKLİĞİ". www.omim.org. Alındı 11 Kasım, 2015.
- ^ a b c d Saini, N (Mart 2015). "Bebeklerde diyabetik ketoasidozu ve septik şoku taklit eden metilmalonik asidemi". Hindistan Yoğun Bakım Tıbbı Dergisi. 19 (3): 183–185. doi:10.4103/0972-5229.152776. PMC 4366921. PMID 25810618.
- ^ Kimberly G Lee. "Yenidoğan tarama testleri". nih.gov. Neonatoloji Bölümü, Güney Karolina Tıp Üniversitesi, Charleston, SC. VeriMed Healthcare Network tarafından sağlanan inceleme. Ayrıca David Zieve, MD, MHA, Isla Ogilvie, PhD ve A.D.A.M. Editör ekibi. Alındı 26 Nisan 2016.
- ^ Coelho D, Suormala T, Stucki M, Lerner-Ellis JP, Rosenblatt DS, Newbold RF, Baumgartner MR, Fowler B (2008). "B vitamini cblD kusuru için gen tanımlama12 metabolizma". N Engl J Med. 358 (14): 1454–64. doi:10.1056 / NEJMoa072200. PMID 18385497.CS1 bakım: birden çok isim: yazarlar listesi (bağlantı)
- ^ Rutsch F, Gailus S, Miousse IR, Suormala T, Sagné C, Toliat MR, Nürnberg G, Wittkampf T, Buers I, Sharifi A, Stucki M, Becker C, Baumgartner M, Robenek H, Marquardt T, Höhne W, Gasnier B , Rosenblatt DS, Fowler B, Nürnberg P (Şubat 2009). "B vitamini cblF kusurunda değiştirilen varsayılan bir lizozomal kobalamin ihracatçısının tanımlanması12 metabolizma". Nat Genet. 41 (2): 234–9. doi:10.1038 / ng.294. PMID 19136951. S2CID 28006539.
- ^ a b c d "OMIM Giriş - # 251000 - METİLMALONİL-KoA MUTAZ YETERSİZLİĞİNE BAĞLI METİLMALONİK ASİDURİ". www.omim.org. Alındı 2015-11-03.
- ^ Oberholzer VG, Levin B, Burgess EA, Young WF (1967). "Metilmalonik asidüri. Kronik metabolik asidoza neden olan doğuştan metabolizma hatası". Arch Dis Çocuk. 42 (225): 492–504. doi:10.1136 / adc.42.225.492. PMC 2019805. PMID 6061291.
- ^ Han, L. (15 Haziran 2015). "Metilmalonik asideminin moleküler mekanizmalarına mikrodizi teknolojisi kullanılarak içgörüler". Uluslararası Klinik ve Deneysel Tıp Dergisi. 8 (6): 8866–8879. PMC 4538064. PMID 26309541. Alındı 5 Kasım 2015.
- ^ Chandler, Randy J. (16 Aralık 2008). "Mut metilmalonik asidemide mitokondriyal disfonksiyon". FASEB Dergisi. 23 (4): 1252–1261. doi:10.1096 / fj.08-121848. PMC 2660647. PMID 19088183.
- ^ Underhill, H (Aralık 2013). "Homozigot MUT mutasyonunda (p.P86L) asemptomatik metilmalonik asidemi". Pediatri Uluslararası. 55 (6): e156-8. doi:10.1111 / ped.12195. PMID 24330302.
daha fazla okuma
- Metilmalonik Asidemi üzerine GeneReviews makale
- GeneReviews Hücre İçi Kobalamin Metabolizması Bozuklukları makalesi
Dış bağlantılar
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |