Morquio sendromu - Morquio syndrome - Wikipedia
| Morquio sendromu | |
|---|---|
| Diğer isimler | Mukopolisakkaridoz IV, MPS IV, Morquio-Brailsford sendromuveya Morquio[1] |
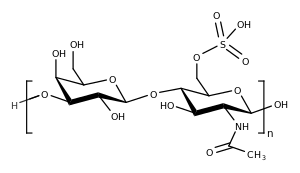 | |
| Keratan sülfat, Morquio sendromlu kişilerin lizozomlarında birikerek semptomlara yol açar. | |
| Uzmanlık | Endokrinoloji |
| Komplikasyonlar | İskelet anormallikleri, işitme kaybı, akciğer yetmezliği, kalp hastalığı |
| Olağan başlangıç | Doğum; durum genellikle 1 ila 3 yaş arasında ortaya çıkar |
| Süresi | Ömür boyu |
| Türler | Tip A ve Tip B |
| Nedenleri | Kalıtsal enzim eksikliği |
| Tedavi | Tip A için elosulfase alfa (Vimizim); Tip B için onaylanmış tedavi yok |
| Prognoz | Azaltılmış ömür. Ölüm genellikle 20-30'lu yaşlarda gerçekleşir |
| Sıklık | 200.000'de 1 ile 300.000'de 1 |
Morquio sendromu, Ayrıca şöyle bilinir Mukopolisakkaridoz Tip IV (MPS IV), nadir metabolik bozukluk Vücudun adı verilen belirli şeker molekülü türlerini işleyemediği glikozaminoglikanlar (AKA GAG'ler veya mukopolisakkaritler). Morquio sendromunda, vücutta oluşan spesifik GAG'ye keratan sülfat. Bu doğum kusuru, hangisi otozomal resesif, bir tür lizozomal depo bozukluğu. GAG'lerin vücudun farklı bölgelerinde birikmesi, birçok farklı organ sisteminde semptomlara neden olur.[2]:544 ABD'de, Morquio sendromunun insidans oranının 200.000'de 1 ile 300.000 canlı doğumda 1 arasında olduğu tahmin edilmektedir.[1][3]
Belirti ve bulgular

Morquio sendromlu hastalar doğumda sağlıklı görünür. Tip A ve B'nin benzer sunumları vardır, ancak Tip B genellikle daha hafif semptomlara sahiptir. Başlangıç yaşı genellikle 1 ila 3 yaş arasındadır. Morquio sendromu, kaburga ve göğüs iskeletinde ilerleyici değişikliklere neden olur ve bu da sinir sıkışması gibi nörolojik komplikasyonlara yol açabilir. Hastalarda ayrıca işitme kaybı ve bulanık kornealar olabilir. Bir hasta tedavi edilmediği sürece zeka genellikle normaldir. hidrosefali.[kaynak belirtilmeli ]
Fiziksel büyüme yavaşlar ve genellikle 8 yaş civarında durur. İskelet anormallikleri arasında çan şeklinde bir göğüs, omurganın düzleşmesi veya eğriliği, kısaltılmış uzun kemikler ve kalça, diz, ayak bilekleri ve bileklerde displazi bulunur. Baş ve boyun arasındaki bağlantıyı stabilize eden kemikler hatalı biçimlendirilebilir (odontoid hipoplazi); bu durumlarda cerrahi prosedür denen spinal servikal kemik füzyonu hayat kurtarıcı olabilir. Kısıtlı nefes alma, eklem sertliği ve kalp hastalığı da yaygındır. MPS IV'ün daha şiddetli formuna sahip çocuklar yirmili veya otuzlu yaşlarının ötesinde yaşayamayabilir.[kaynak belirtilmeli ]
Sebep olmak
Morquio sendromu, bir otozomal resesif kalıtsal gen. Her insan, keratan sülfatı parçalamak için gereken genlerin iki kopyasına sahiptir, ancak yalnızca bir sağlıklı kopyaya ihtiyaç vardır. Her iki ebeveyn de çocuklarına kusurlu bir kopya verir ve bu, genin işlevsel kopyası olmayan bir çocukla sonuçlanır. Bu nedenle vücut, atılmak üzere keratan sülfatı parçalayamaz. Tamamen parçalanmış GAG'ler vücuttaki hücrelerde depolanarak ilerleyici hasara neden olur. Bebekler hastalığın çok az belirtisini gösterebilir, ancak giderek daha fazla hücre hasar gördükçe semptomlar ortaya çıkmaya başlar.[4]
Teşhis
Sınıflandırma
Bu sendromun Morquio A ve Morquio B sendromu veya MPS IVA ve MPS IVB olarak adlandırılan A ve B olmak üzere iki formu vardır. İki form, ilgili gen ürününe göre ayırt edilir; Tip A, cihazdaki bir arızayı içerir. GALNS Gen, B Tipi ise GLB1 gen.[kaynak belirtilmeli ]
| Morquio sendromu tipi | Gen | Eksik enzim | Kromozomal bölge |
|---|---|---|---|
| A yazın | GALNS | Galaktozamin-6 sülfataz | 16q24 |
| B Tipi | GLB1 | Beta-galaktosidaz | 3p22 |
Tedavi
Morquio sendromunun tedavisi şunlardan oluşur: doğum öncesi kimlik ve enzim replasman tedavisi. 12 Şubat 2014 tarihinde ABD Gıda ve İlaç İdaresi ilacı onayladı elosülfaz alfa Tip A tedavisi için (Vimizim) Şu anda Tip B için bir tedavi yoktur.[4]
Prognoz
Morquio sendromlu hastaların yaşam süresi değişkendir ve alt tipe bağlıdır. Tip A, 20'li ve 30'lu yaşlarda bir yaşam beklentisiyle genellikle şiddetlidir. [5] 2016 yılında Morquio sendromlu bir adam 81 yaşında öldü.[6]
Bir çalışma, hastanedeki hastalar için ortalama yaşam beklentisinin Birleşik Krallık 25.30'du standart sapma 17.43 yıl. Ortalama olarak, kadın hastalar erkek hastalardan 4 yıl daha uzun yaşadı. Solunum yetmezliği hastaların% 63'ünde birincil ölüm nedeniydi. Diğer ölüm nedenleri kalp yetmezliği (% 11), travma sonrası organ yetmezliği (% 11), ameliyatın komplikasyonları (% 11) ve kalp krizi (% 4). Yaşam beklentisi 1980'lerden beri artıyor. Solunum yetmezliğine bağlı ortalama ölüm yaşı 1980'lerde 17,42'den 2000'lerde 30,74'e yükseldi.[7]
Tarih
Durum ilk olarak 1929'da eş zamanlı ve bağımsız olarak Luis Morquio (1867–1935),[8] öne çıkan Uruguaylı içinde keşfeden doktor Montevideo, ve James Frederick Brailsford (1888–1961), bir İngiliz radyolog Birmingham, İngiltere.[9][10] İkisi de olayının farkına vardı kornea bulutlanma aort kapak hastalığı ve keratan sülfatın idrarla atılımı. Morquio, İsveç kökenli bir ailede dört kardeşte görülen rahatsızlığı gözlemledi ve gözlemlerini Fransızca olarak bildirdi.[kaynak belirtilmeli ]
Ayrıca bakınız
- Hurler sendromu (MPS I )
- Hunter sendromu (MPS II)
- Sanfilippo sendromu (MPS III)
- Cücelik
Referanslar
- ^ a b "MPS IV (Morquio sendromu)". MPSSociety.org. Ulusal MPS Topluluğu. Arşivlenen orijinal 21 Ağustos 2017. Alındı 14 Ocak 2015.
- ^ James, William D .; Berger, Timothy G. (2006). Andrews'un Deri Hastalıkları: klinik Dermatoloji. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ^ Prat C, Lemaire O, Bret J, Zabraniecki L, Fournié B (Mayıs 2008). "Morquio sendromu: Bir yetişkinde tanı". Eklem Kemik Omurga. 75 (4): 495–8. doi:10.1016 / j.jbspin.2007.07.021. PMID 18456538.
- ^ a b "MPS IV (Morquio Sendromu)". Canadian MPS Society. Alındı 14 Haziran 2019.
- ^ "Mukopolisakkaridozlar Bilgi Sayfası". Ulusal Nörolojik Bozukluklar ve İnme Enstitüsü. 13 Mayıs 2019. Alındı 14 Haziran 2019.
- ^ Blacketer, Rosie (23 Eylül 2016). "Kenneth Dean Martin". Osage County Herald-Chronicle. Alındı 14 Haziran 2019.
- ^ Lavery, Christine; Hendriksz, Chris (10 Nisan 2014). "Morquio Sendromu A Hastalarında Ölüm". Kalıtsal Metabolik Hastalık Dergisi. JIMD Raporları. 15: 59–66. doi:10.1007/8904_2014_298. PMC 4270860. PMID 24718838.
- ^ Morquio, L. (1929). "Sur une forme de distrophie osseuse familiale". Archives de Médecine des Infants. Paris. 32: 129–135. ISSN 0365-4311.
- ^ synd / 2108 -de Kim Adlandırdı?
- ^ Brailsford, J.F. (1929). "Kondro-osteo-distrofi: Omur çıkığı olan bir çocuğun röntgenografik ve klinik özellikleri". American Journal of Surgery. New York. 7 (3): 404–410. doi:10.1016 / S0002-9610 (29) 90496-7.
Dış bağlantılar
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |