Oksoamonyum katalizli oksidasyon - Oxoammonium-catalyzed oxidation
Bu makale çok güveniyor Referanslar -e birincil kaynaklar. (Nisan 2019) (Bu şablon mesajını nasıl ve ne zaman kaldıracağınızı öğrenin) |
Oksoamonyum ile katalize edilen oksidasyon reaksiyonları dahil etmek dönüştürmek nın-nin organik substratlar daha çok oksitlenmiş bir eylem yoluyla malzemeler N-oksoamonyum türleri. Nitroksitler, stokiyometrik miktarda bir terminal oksidan varlığında katalitik miktarlarda da kullanılabilir.[1] Kullanılan nitroksit radikal türleri, 2,2,6,6-tetrametilpiperidin-1-oksil (TEMPO) veya bunların türevleridir.
(1)

Mekanizma ve stereokimya
Nitroksitin bir elektron oksidasyonu, aktif oksitleyici ajan olarak görev yapan oldukça elektrofilik bir oksoamonyum türü üretir.[2] Nitroksit, daha ucuz stoikiometrik oksidanlar gibi bir katalizör olarak kullanılabilir. sodyum hipoklorit[3] veya bis (asetoksi) iyodobenzen (BAIB).[4]
Nötr veya hafif asidik koşullar altında (örneğin, silika jel varlığında), oksidasyon, hidroksil grubu ile oksoamonyum nitrojen arasındaki bir başlangıç hidrojen bağı, ardından uyumlu proton transferi ve hidrit soyutlaması ile meydana gelir.[5] Hidrojen bağına duyulan ihtiyaç, rekabet gücü yüksek β-alkoksi ve β-amino alkollerin düşük reaktivitesiyle desteklenir. moleküliçi hidrojen bağı. Zayıf bazik (piridin) koşullar altında oksidasyon mekanizması benzerdir, ancak piridin hidroksiamonyum türlerini nötralize eder ve bu ara ürün, nitroksit radikalleri ve piridinyum tuzları vermek için oksoamonyum tuzu ile "eşlik eder" (aşağıdaki denklem (3) 'e bakınız). Bu reaksiyon baz ve aktif oksidan tükettiği için, zayıf bazik koşullar altında iki eşdeğer baz ve oksidan gereklidir. Yakın tarihli bir makalede sunulan tarafsız ve temel koşullar altında birleşik bir mekanizma.[6] Yazarlar, bir dizi oksoamonyum tuzu aracılı oksidasyonların kapsamlı bir analizini sunar.
(2)

Güçlü bazik koşullar altında protondan arındırılmış substrat N-oksamonyum türleri ile reaksiyona girer. Substrat alkoksidin nitrojen veya oksijene saldırması meydana gelebilir, ancak ilki N-alkoksi aminlerin oksidasyonlarının gözlemlerine dayalı olarak çalıştığına inanılmaktadır (ki bu muhtemelen ara madde yoluyla ilerler). 1).[7] İndirgenmiş ürünün (bir hidroksilamin) oksoamonyum iyonu ile karşılaştırılması oksidasyon ile rekabet eder; bu nedenle, genellikle fazla miktarda oksitleyici ajan gerekir.
(3)

Nitroksitle katalize edilen oksidasyonlar, aktif oksitleyici ajan olarak N-oksoamonyum ara ürünlerini içerir. Nitroksit radikalinin oksidasyon mekanizması, kullanılan terminal oksidana bağlıdır. NaOCl gibi iki elektronlu oksidanlar, nitroksitleri doğrudan oksoamonyuma dönüştürebilir.
(4)

Bakır (II) gibi tek elektronlu oksidanlar, terminal oksidan olarak dioksijen içeren daha karmaşık bir mekanizma yoluyla çalışır.[8] Bakır (II), dört eşdeğer nitroksidi oksoamonyuma oksitler, iki eşdeğeri (mavi) karbonil bileşikleri oluşturmak için alkollerle reaksiyona girer. Diğer iki eşdeğer oksoamonyum (kırmızı), nitroksi radikallerini (pembe) yeniden oluşturmak için orantılı hale getirilir. Son olarak, dioksijen, dört eşdeğer bakır (I) 'i tekrar bakır (II)' ye yeniden okside eder. Genel olarak, tek bir dioksijen molekülü, iki eşdeğer su oluşumu ile iki eşdeğer alkolün oksidasyonuna aracılık eder.
(5)

Stereoselektif varyantlar
Enantiyoselektif oksidasyonlar tipik olarak kiral alkollerin kinetik çözünürlükleridir veya desimetrizasyon reaksiyonlarıdır. Bu oksidasyonlar, katalitik modda kiral nitroksit radikallerinin kullanılmasıyla kolaylaştırılabilir. Rasemik 1-feniletanolün kinetik çözünürlüğüyle iyi bir örnek sağlanır.[9] Öte yandan, oksoamonyum oksidanların kullanıldığı oksidatif desimetrizasyon işlemleri nispeten nadirdir.[10]
(6)

Dürbün
Oksoamonyum tuzlarının kullanıldığı oksidasyonlar, asidik veya bazik koşullar altında stoikiometrik veya katalitik modda gerçekleştirilebilir. Bu bölüm, alkollerin oksoamonyum tuzları ile karbonil bileşiklere stoikiometrik ve katalitik oksidasyonu için en yaygın olarak kullanılan koşulları açıklamaktadır. TEMPO kullanılarak çok çeşitli alkoller oksitlenebilse de, bazen daha fazla elektron açısından zengin işlevselliğin rekabetçi oksidasyonu gerçekleşir. Ek olarak, poliollerin oksidasyonunun saha seçiciliği, kullanılan koşullara bağlı olarak değişebilir.
Stokiyometrik oksidasyonlar
Hafif asidik veya nötr koşullar altında, oksoamonyum tuzları, örneğin Bobbitt tuzu alilik, benzilik oksitlemek,[11] propargylic,[12] veya alifatik alkolleri karşılık gelen aldehitlere veya ketonlara dönüştürür. Seçicilik düşük olmasına rağmen ikincil alkoller birincil alkollerden daha hızlı reaksiyona girer. Uygun bir deneysel protokol, oksoamonyum tuzunun geri dönüşümüne izin verir.[12]
(7)

Aminler, benzilik eterler ve alkenler, aktive edilmemiş alkollerden daha hızlı oksitlenir; bu nedenle, bu fonksiyonel grupların varlığında aktifleştirilmemiş alkollerin seçici stoikiometrik oksidasyonu mümkün değildir.[13] Β-nitrojen veya β-oksijen ikameli alkoller, asidik koşullar altında yavaş bir şekilde reaksiyona girer.[12] Alilik ve benzilik alkoller, bu koşullar altında seçici olarak oksitlenebilir.[13]
(8)

Temel koşullar altında, indirgenmiş nitroksit ve reaksiyona girmemiş oksoamonyum arasındaki rekabetçi orantıdan dolayı iki eşdeğer oksidan gereklidir (yukarıdaki denklem (3) 'e bakınız). Piridin genellikle baz olarak kullanılır. Bunlar stokiyometrik modda nitroksit oksidasyonları için en yaygın koşullardır.
(9)

Katalitik oksidasyonlar
Katalitik oksoamonyum oksidasyonu, terminal oksidan olarak sodyum hipoklorit kullanılarak kolaylaştırılabilir. Reaksiyonun ilerlemesi için bir tampon kullanılarak pH 10'un altında tutulmalıdır. Nitroksitin aktif oksitleyici maddesi hipobromit anyondur; dolayısıyla potasyum bromür katkı maddesi olarak kullanılır.[3] Karbonil içeren ürünlerde a-stereojenik merkezlerin epimerizasyonu gerçekleşmez.
(10)

Kloritlerin hem hipokloritler hem de TEMPO ile birlikte terminal oksidanlar olarak kullanılması, klorlama yan ürünleri içermeyen karboksilik asitler verir.[14] Reaksiyon genellikle aynı potada iki aşamada gerçekleştirilir: TEMPO ve hipoklorit ile kısmi oksidasyon gerçekleştirilir, ardından oksidasyonu tamamlamak için klorit eklenir. Yalnızca birincil alkol oksidasyonu gözlenir. Sharpless dihidroksilasyon ile bağlantılı olarak, bu yöntem enantiyopür α-hidroksi asitleri oluşturmak için kullanılabilir.[15]
(11)

Yukarıdaki yöntemlerin her ikisinin de önemli bir sınırlaması, her ikisi de rekabetçi oksidasyona maruz kalan serbest amin veya alken işlevselliği ile uyumsuzluktur. Terminal oksidan olarak bis (asetoksi) iyodobenzen (BAIB) kullanılması bu sorunu ortadan kaldırır. BAIB, nitroksit radikalini doğrudan oksitleyemez ve ilk oksoamonyum oluşumunun asitle katalize edilen orantısızlıktan kaynaklandığına inanılmaktadır. BAIB daha sonra elde edilen hidroksilamini bir oksoamonyum tuzuna oksitleyebilir. Reaksiyon asidik koşullar altında gerçekleştirilse de (asetik asit bir yan üründür ve orantısızlığı kolaylaştırmak için sıklıkla eklenir), birincil alkol oksidasyonu için seçicilik önemlidir.[4] Epoksitler gibi baza duyarlı fonksiyonel gruplar, bu koşullar altında tolere edilir.[16]
(12)
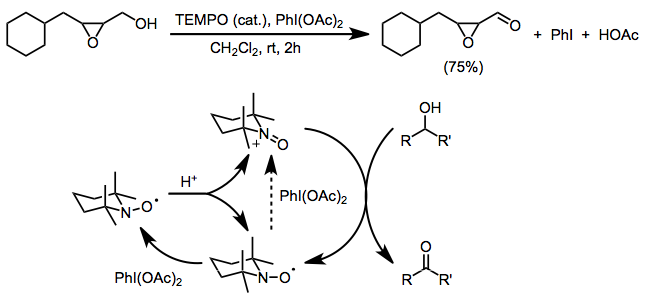
TEMPO ile kullanılan diğer iki elektronlu terminal oksidanlar arasında mCPBA (ikincil oksidasyon tercih edilir, ancak yan reaksiyonlar meydana gelebilir),[17] N-klorosüksinimid,[18] ve Oxone.[19]
Bakır (II), hem serbest klorür tuzu hem de iki dişli ligandlı bir kompleks olarak TEMPO'yu oksoamonyum tuzuna oksitlemektedir. Bu reaksiyonlarda hava, son oksidan görevi görür.[20] Havanın bakır (I) 'i bakıra (II) oksitleyip oksitlemediği veya alkol oksidasyonunun kısmen bakırın aracılık edip etmediği ve havanın ortaya çıkan hidroksilamini oksoamonyum tuzuna geri oksitleyip yükseltmediği açık değildir. İlki, Wacker süreci ancak ikincisi, bakır komplekslerinin ve diğer birkaç metal kompleksinin neden TEMPO ile birlikte alkolleri oksitleyebildiğini açıklıyor.
(13)

Aktif manganez dioksit Alilik ve benzilik alkolleri oksitleyen, TEMPO'dan daha ucuzdur ve operasyonel olarak kullanımı kolaydır.[21] Gibi krom bazlı reaktifler piridinyum klorokromat alkolleri karbonil bileşiklere dönüştürmek için de kullanılabilir; krom atıklarının stokiyometrik oluşumu bir dezavantaj olmasına rağmen.[22] Kullanılan oksidasyonlar dimetil sülfoksit, benzeri Swern ve Moffatt reaksiyonlar, ağır metaller içermez ve çok çeşitli substratları okside eder.[23] Oksoamonyum oksidasyonları, diollerin ve asetilenik alkollerin reaksiyonları için DMSO yöntemlerine tercih edilir. Dess-Martin periodinane birincil dezavantajları hazırlama ve güvenlik güçlükleri olan alkollerin oldukça seçici, hafif bir oksidanıdır.[24]
Referanslar
- ^ Bobbitt, J. M.; Bruckner, C .; Merbouh, N. Org. Tepki. 2009, 74, 103. doi:10.1002 / 0471264180.or074.02
- ^ Merbouh, N .; Bobbitt, J. M .; Brückner, C. J. Org. Chem. 2004, 69, 5116.
- ^ a b Sheldon, R.A.; Arends, I.W.C. E .; ten Brink, G. J .; Dijksman, A. Acc. Chem. Res. 2002, 35, 774. doi:10.1021 / ar010075n
- ^ a b De Mico, A .; Margarita, R .; Parlanti, L .; Vescovi, A .; Piancatelli, G. J. Org. Chem. 1997, 62, 6974.
- ^ Bailey, W. F .; Bobbitt, J. M .; Wiberg, K. B. J. Org. Chem. 2007, 72, 4504.
- ^ Hamlin, T. A .; Kelly, C. B .; Ovian, J. M .; Wiles, R. J .; Tilley, L. J .; Kurşun çırpıcı, N.E. J. Org. Chem. 2015, 80, 8150.
- ^ Semmelhack, M. F .; Schmid, C. R .; Cortés, D. A. Tetrahedron Lett. 1986, 27, 1119.
- ^ Semmelhack, M. F .; Schmid, C. R .; Cortés, D. A .; Chou, C. S. J. Am. Chem. Soc. 1984, 106, 3374.
- ^ Rychnovsky, S. D .; McLernon, T. L .; Rajapakse, H. J. Org. Chem. 1996, 61, 1194.
- ^ Tanaka, H .; Kawakami, Y .; Goto, K .; Kuroboshi, M. Tetrahedron Lett. 2001, 42, 445.
- ^ Miyazawa, T .; Endo, T .; Shiihashi, S .; Okawara, M. J. Org. Chem. 1985, 50, 1332.
- ^ a b c Bobbitt, J. M. J. Org. Chem. 1998, 63, 9367.
- ^ a b Bobbitt, J. M .; Merbouh, N. Org. Synth. 2005, 82, 80.>
- ^ Şarkı, Z. J .; Zhao, M .; Desmond, R .; Devine, P .; Tschaen, D. M .; Tillyer, R .; Frey, L .; Heid, R .; Xu, F .; Foster, B .; Li, J .; Reamer, R .; Volante, R .; Grabowski, E. J. J .; Dolling, U. H .; Reider, P. J .; Okada, S .; Kato, Y .; Mano, E. J. Org. Chem. 1999, 64, 9658.
- ^ Sharpless, K. B .; Amberg, W .; Bennani, Y. L .; Crispino, G. A .; Hartung, J .; Jeong, K. S .; Kwong, H.L .; Morikawa, K .; Wang, Z. M .; Xu, D .; Zhang, X. L. J. Org. Chem. 1992, 57, 2768.
- ^ De Mico, A .; Margarita, R .; Parlanti, L .; Vescovi, A .; Piancatelli, G. J. Org. Chem. 1997, 62, 6974.
- ^ Ganem, B. J. Org. Chem. 1975, 40, 1998.
- ^ Einhorn, J .; Einhorn, C .; Ratajczak, F .; Pierre, J.-L. J. Org. Chem. 1996, 61, 7452.
- ^ Bolm, C .; Magnus, A. S .; Hildebrand, J. P. Org. Lett. 2000, 2, 1173.
- ^ Sheldon, R.A.; Arends, I.W.C. E. Adv. Synth. Katal. 2004, 346, 1051.
- ^ Taylor, R. J. K .; Reid, M .; Foot, J .; Raw, S.A. Acc. Chem. Res. 2005, 38, 851.
- ^ Luzzio, F.A. Org. Tepki. 1998, 53, 1.
- ^ Tidwell, T. T. Org. Tepki. 1990, 39, 297.
- ^ Dess, D. B .; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277.