Kinetik izotop etkisi - Kinetic isotope effect
![{ displaystyle { begin {matrix} { ce {{CN ^ {-}} + {^ {12} CH3-Br} -> [k_ {12}] {^ {12} CH3-CN} + Br ^ {-}}} { ce {{CN ^ {-}} + {^ {13} CH3-Br} -> [k_ {13}] {^ {13} CH3-CN} + Br ^ {-}}} {} end {matrix}} qquad { text {KIE}} = { frac {k_ {12}} {k_ {13}}} = 1.082 pm 0.008}](https://wikimedia.org/api/rest_v1/media/math/render/svg/438109ea220fd190ccc57f3e2c3726c47c24aae0)
Tepkisinde metil bromür ile siyanür,
karbonun kinetik izotop etkisi metil grubu 1.082 ± 0.008 olarak bulundu.[1][2]
İçinde fiziksel organik kimya, bir kinetik izotop etkisi (KIE) değişikliktir reaksiyon hızı bir Kimyasal reaksiyon ne zaman atomlar içinde reaktanlar biriyle değiştirilir izotoplar.[3] Resmi olarak, oranıdır hız sabitleri ışığı içeren tepkiler için (kL) ve ağır (kH) izotopik olarak ikame edilmiş reaktanlar (izotopologlar):

Reaksiyon hızındaki bu değişiklik, öncelikle daha ağır olmasından kaynaklanan kuantum mekanik bir etkidir. izotopologlar daha düşük olan titreşim daha hafif meslektaşlarına kıyasla frekanslar. Çoğu durumda, bu, daha ağır izotopologların hedefe ulaşması için gereken daha büyük bir enerji girdisi anlamına gelir geçiş durumu (veya nadir durumlarda, ayrışma sınırı ) ve sonuç olarak daha yavaş bir reaksiyon hızı. Kinetik izotop etkilerinin incelenmesi, reaksiyon mekanizması bazı kimyasal reaksiyonların ve bazen ilaç geliştirmede olumsuzlukları iyileştirmek için istismar edilmektedir. farmakokinetik metabolik olarak savunmasız C-H bağlarını koruyarak.
Arka fon
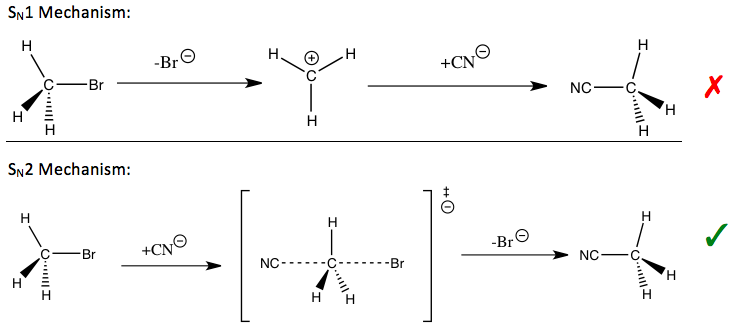
Kinetik izotop etkisi, çalışma için en önemli ve hassas araçlardan biri olarak kabul edilir. reaksiyon mekanizmaları bilgisi, karşılık gelen reaksiyonların arzu edilen niteliklerinin geliştirilmesine izin verir. Örneğin, kinetik izotop etkileri, bir nükleofilik ikame reaksiyon bir tek moleküllü (SN1) veya iki moleküllü (SN2) yol.
Tepkisinde metil bromür ve siyanür (girişte gösterilmiştir), gözlemlenen metil karbon kinetik izotop etkisi, bir SN2 mekanizma.[1] Yola bağlı olarak, stabilize etmek için farklı stratejiler kullanılabilir. geçiş durumu of oran belirleme adımı reaksiyonun ve iyileştirmenin reaksiyon hızı ve endüstriyel uygulamalar için önemli olan seçicilik.
İzotopik hız değişiklikleri en çok göreceli kitle etki, etkilenen bağların titreşim frekansları ile ilgili olduğundan, değişim en büyüktür. Örneğin, bir hidrojen atom (H) 'yi izotopuna döteryum (D) kütlede% 100 artışı temsil ederken, değiştirirken karbon Karbon-13 ile -12, kütle yalnızca yüzde 8 artar. Bir C – H bağı içeren bir reaksiyonun hızı tipik olarak karşılık gelen C – D bağından 6–10 kat daha hızlıdır. 12C reaksiyonu, karşılık gelen reaksiyondan yalnızca yüzde 4 daha hızlıdır. 13C reaksiyonu[4]:445 (her iki durumda da izotop bir Atomik kütle birimi daha ağır).
İzotopik ikame, reaksiyon hızını çeşitli şekillerde değiştirebilir. Pek çok durumda, hız farkı, bir atomun kütlesinin onu etkilediğine dikkat edilerek rasyonelleştirilebilir. titreşim frekansı of Kimyasal bağ oluştursa bile potansiyel enerji yüzeyi çünkü reaksiyon neredeyse aynı. Daha ağır izotoplar (klasik olarak ) daha düşük titreşim frekanslarına yol açar veya kuantum mekanik olarak, daha düşük olacak sıfır nokta enerjisi. Daha düşük sıfır noktası enerjisi ile, bağı koparmak için daha fazla enerji sağlanmalıdır, bu da daha yüksek aktivasyon enerjisi bağ bölünmesi için, bu da ölçülen oranı düşürür (bkz., örneğin, Arrhenius denklemi ).[3][4]:427
Sınıflandırma
Birincil kinetik izotop etkileri
Bir birincil kinetik izotop etkisi izotopik olarak etiketlenmiş atoma bir bağ oluştuğunda veya kırıldığında bulunabilir.[3][4]:427 Bir kinetik izotop etkisinin araştırılma şekline bağlı olarak (hızların paralel ölçümü vs. moleküller arası rekabet vs. Molekül içi rekabet), bir birincil kinetik izotop etkisinin gözlemlenmesi, hız sınırlama aşamasında veya sonraki ürün belirleme aşamalarında izotopa bir bağın kopması / oluşturulmasının göstergesidir. (Birincil kinetik izotop etkisinin, hız sınırlayıcı adımda izotopa bağ bölünmesini / oluşumunu yansıtması gerektiği yanılgısı, ders kitaplarında ve birincil literatürde sıklıkla tekrarlanır: bölüme bakın deneyler altında.)[5]
Daha önce bahsedilen nükleofilik ikame reaksiyonları için, ikamenin meydana geldiği hem ayrılan gruplar, hem nükleofiller hem de a-karbon için birincil kinetik izotop etkileri araştırılmıştır. Ayrılan grup kinetik izotop etkilerinin yorumlanması, sıcaklıktan bağımsız faktörlerin önemli katkıları nedeniyle ilk başta zor olmuştur. Α-karbondaki kinetik izotop etkileri, S'deki geçiş durumunun simetrisini anlamak için kullanılabilir.N2 reaksiyon, bu kinetik izotop etkisi ideal olandan daha az hassas olsa da, titreşim dışı faktörlerin de katkısı nedeniyle.[1]
İkincil kinetik izotop etkileri
Bir ikincil kinetik izotop etkisi reaktantta izotopik olarak etiketlenmiş atoma hiçbir bağ kopmadığında veya oluşmadığında gözlemlenir.[3][4]:427 İkincil kinetik izotop etkileri, birincil kinetik izotop etkilerinden çok daha küçük olma eğilimindedir; ancak ikincil döteryum izotop etkileri döteryum atomu başına 1.4 kadar büyük olabilir ve ağır element izotop etkilerini çok yüksek hassasiyette ölçmek için teknikler geliştirilmiştir, bu nedenle ikincil kinetik izotop etkileri, reaksiyon mekanizmalarını aydınlatmak için hala çok yararlıdır.
Yukarıda belirtilen nükleofilik ikame reaksiyonları için, α-karbondaki ikincil hidrojen kinetik izotop etkileri, S'yi ayırt etmek için doğrudan bir yol sağlar.N1 ve SN2 reaksiyon. S olduğu bulunmuşturN1 reaksiyon tipik olarak büyük ikincil kinetik izotop etkilerine yol açar, teorik maksimumlarına yaklaşık 1.22'ye yaklaşırken, SN2 reaksiyon tipik olarak, birliğe çok yakın veya daha az birincil kinetik izotop etkileri verir. 1'den büyük olan kinetik izotop etkilerine normal kinetik izotop etkileribirden az kinetik izotop etkileri olarak anılırken ters kinetik izotop etkileri. Genel olarak, geçiş durumundaki daha küçük kuvvet sabitlerinin normal bir kinetik izotop etkisi vermesi beklenir ve geçiş durumundaki daha büyük kuvvet sabitlerinin, gerilme titreşimli katkıları kinetik izotop etkisine baskın olduğunda ters kinetik izotop etkisi vermesi beklenir.[1]
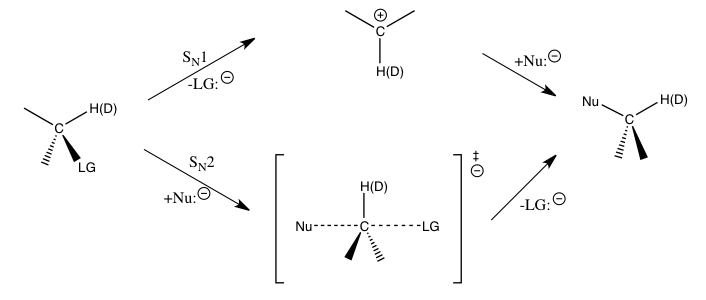
Α-karbondaki bu tür ikincil izotop etkilerinin büyüklükleri büyük ölçüde Cα-H (D) titreşimler. S içinN1 reaksiyon, çünkü karbon bir sp'ye dönüştürüldüğünden2 C'deki bir artışla hız belirleme adımı için geçiş durumu sırasında hibridize karbenium iyonuα-H (D) bağ düzeninde, sadece gerilme titreşimleri önemli olsaydı ters kinetik izotop etkisi beklenirdi. Gözlemlenen büyük normal kinetik izotop etkilerinin, reaktanlardan karbenium oluşumunun geçiş durumuna geçerken düzlemden önemli bükülme titreşim katkılarından kaynaklandığı bulunmuştur. S içinN2 reaksiyon, bükülme titreşimleri kinetik izotop etkisi için hala önemli bir rol oynamaktadır, ancak gerilme titreşimi katkıları daha benzer büyüklüktedir ve ortaya çıkan kinetik izotop etkisi, ilgili titreşimlerin spesifik katkılarına bağlı olarak normal veya ters olabilir.[1][6][7]
Teori
İzotop etkilerinin teorik tedavisi büyük ölçüde şunlara dayanır: geçiş durumu teorisi Bu, reaksiyon için tek bir potansiyel enerji yüzeyini ve tepkimeye giren maddeler ile bu yüzeydeki ürünler arasında, üstte geçiş halinin bulunduğu bir bariyer olduğunu varsayar.[8][9] Kinetik izotop etkisi, büyük ölçüde, potansiyel enerji yüzeyinin minimum enerji yolu boyunca izotopik pertürbasyon tarafından üretilen titreşimsel zemin durumlarındaki değişikliklerden kaynaklanır; bu, yalnızca sistemin kuantum mekanik işlemleriyle açıklanabilir. Reaksiyon koordinatı boyunca hareket eden atomun kütlesine ve enerji bariyerinin yapısına (genişlik ve yükseklik) bağlı olarak, kuantum mekanik tünelleme ayrıca, gözlemlenen bir kinetik izotop etkisine büyük bir katkıda bulunabilir ve "yarı klasik" geçiş durumu teorisi modeline ek olarak ayrı ayrı ele alınması gerekebilir.[8]
Döteryum kinetik izotop etkisi (2H KIE), en yaygın, kullanışlı ve iyi anlaşılmış kinetik izotop etkisidir. Yoğunluk fonksiyonel teorisi hesaplamaları kullanılarak bir döteryum kinetik izotop etkisinin sayısal değerinin doğru tahmini artık nispeten rutindir. Dahası, birkaç kalitatif ve yarı-kantitatif model, döteryum izotop etkilerinin kaba tahminlerinin hesaplamalar olmadan yapılmasına izin verir, genellikle deneysel verileri rasyonelleştirmek veya hatta farklı mekanik olasılıkları desteklemek veya çürütmek için yeterli bilgi sağlar. Döteryum içeren başlangıç malzemeleri genellikle ticari olarak temin edilebilir, bu da izotopik olarak zenginleştirilmiş başlangıç malzemelerinin sentezini nispeten basit hale getirir. Ayrıca döteryum ve protium kütlesindeki büyük nispi fark ve titreşim frekanslarındaki buna bağlı farklılıklar nedeniyle, izotop etkisinin büyüklüğü, protium ve trityum dışındaki diğer izotop çiftlerinden daha büyüktür,[10] hem birincil hem de ikincil izotop etkilerinin kolayca ölçülmesine ve yorumlanmasına izin verir. Aksine, ikincil etkiler daha ağır elementler için genellikle çok küçüktür ve büyüklük olarak deneysel belirsizliğe yakındır, bu da yorumlanmasını zorlaştırır ve kullanımlarını sınırlar. İzotop etkileri bağlamında, hidrojen genellikle hafif izotop, protium (1H), özellikle. Bu makalenin geri kalanında, hidrojen ve döteryum paralel gramer yapıları veya aralarındaki doğrudan karşılaştırmalar, protium ve döteryum'a atıfta bulunarak yorumlanmalıdır.[11]
Kinetik izotop etkileri teorisi ilk olarak şu şekilde formüle edildi: Jacob Bigeleisen 1949'da.[12][4]:427 Bigeleisen'in döteryum kinetik izotop etkileri için genel formülü (daha ağır elementler için de geçerlidir) aşağıda verilmiştir. Hız sabitlerinin hesaplanması için geçiş durumu teorisini ve öteleme, dönme ve titreşim seviyelerinin istatistiksel mekanik bir muamelesini kullanır. kH ve kD. Bununla birlikte, bu formül, genellikle ayrı bir düzeltme faktörü olarak tanıtılan kuantum tünellemeden gelen katkıyı ihmal ettiği için "yarı klasiktir". Bigeleisen'in formülü, bir C – H bağına kıyasla biraz daha kısa olan C – D bağının neden olduğu bağlı olmayan itici etkileşimlerdeki farklılıklarla da ilgilenmez. Denklemde, H veya D alt simgeli miktarlar sırasıyla hidrojen veya döteryum ikameli türlere atıfta bulunurken, çift hançerli veya kancasız miktarlar, sırasıyla geçiş durumuna veya reaktan temel durumuna karşılık gelir.[7][13] (Kesinlikle, a iletim katsayılarındaki izotopik bir farktan kaynaklanan terim de dahil edilmelidir.[14])

- ,
![{ displaystyle { frac {k _ {{ ce {H}}}} {k _ {{ ce {D}}}}} = sol ({ frac { sigma _ {{ ce {H}} } sigma _ {{ ce {D}}} ^ { ddagger}} { sigma _ {{ ce {D}}} sigma _ {{ ce {H}}} ^ { ddagger}} } sağ) left ({ frac {M _ {{ ce {H}}} ^ { ddagger} M _ {{ ce {D}}}} {M _ {{ ce {D}}} ^ { ddagger} M _ {{ ce {H}}}}} sağ) ^ { frac {3} {2}} left ({ frac {I_ {x { ce {H}}} ^ { ddagger} I_ {y { ce {H}}} ^ { ddagger} I_ {z { ce {H}}} ^ { ddagger}} {I_ {x { ce {D}}} ^ { ddagger} I_ {y { ce {D}}} ^ { ddagger} I_ {z { ce {D}}} ^ { ddagger}}} { frac {I_ {x { ce {D}} } I_ {y { ce {D}}} I_ {z { ce {D}}}} {I_ {x { ce {H}}} I_ {y { ce {H}}} I_ {z { ce {H}}}}} sağ) ^ { frac {1} {2}} left ({ frac { prod limits _ {i = 1} ^ {3N ^ { ddagger} - 7} { frac {1-e ^ {- u_ {i { ce {D}}} ^ { ddagger}}} {1-e ^ {- u_ {i { ce {H}}} ^ { ddagger}}}}} { prod limits _ {i = 1} ^ {3N-6} { frac {1-e ^ {- u_ {i { ce {D}}}}} {1- e ^ {- u_ {i { ce {H}}}}}}} sağ) e ^ {- { frac {1} {2}} left [ sum limits _ {i = 1} ^ {3N ^ { ddagger} -7} (u_ {i { ce {H}}} ^ { ddagger} -u_ {i { ce {D}}} ^ { ddagger}) - toplam limitler _ {i = 1} ^ {3N-6} (u_ {i { ce {H}}} - u_ {i { ce {D}}}) sağ]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/93f26faede9d0fba35d6f675e641c716e7284c0d)
nerede tanımlıyoruz
- ve .


Buraya, h ... Planck sabiti, kB ... Boltzmann sabiti, olarak ifade edilen titreşim frekansıdır wavenumbers, c ... ışık hızı, NBir ... Avogadro sabiti, ve R ... Evrensel gaz sabiti. ΣX (X = H veya D), reaktanlar ve geçiş durumları için simetri sayılarıdır. MX karşılık gelen türlerin moleküler kütleleri ve benqX (q = x, yveya z) terimler, üç ana eksenle ilgili eylemsizlik momentleridir. senbenX karşılık gelen titreşim frekansları ile doğru orantılıdır, νbenve titreşimsel sıfır nokta enerjisi (aşağıya bakınız). Tamsayılar N ve N‡ sırasıyla reaktanlardaki ve geçiş durumlarındaki atom sayısıdır.[7] Yukarıda verilen karmaşık ifade, dört ayrı faktörün ürünü olarak gösterilebilir:[7]

- .

Döteryum izotop etkilerinin özel durumu için, ilk üç terimin birliğe eşit veya birliğe çok yakın olarak değerlendirilebileceğini savunacağız. İlk faktör S (σ içerenX) çeşitli türler için simetri sayılarının oranıdır. Bu, reaktanlardaki ve geçiş halindeki özdeş atomların veya grupların permütasyonuna yol açan moleküler ve bağ dönüşlerinin sayısına bağlı olan rasyonel bir sayı (tam sayıların oranı) olacaktır.[13] Düşük simetriye sahip sistemler için tüm σX (reaktan ve geçiş durumu) birlik olacaktır; Böylece S genellikle ihmal edilebilir. MMI faktör (içeren MX ve benqX) moleküler kütlelerin oranını ve atalet momentlerini ifade eder. Hidrojen ve döteryum, çoğu reaktan ve geçiş durumuna kıyasla çok daha hafif olma eğiliminde olduğundan, H ve D içeren moleküller arasındaki moleküler kütleler ve eylemsizlik momentleri arasında çok az fark vardır. MMI faktör genellikle birlik olarak da tahmin edilir. EXC faktör (titreşim ürününü içeren bölüm fonksiyonları ) titreşimle uyarılmış moleküllerin reaksiyonlarının neden olduğu kinetik izotop etkisini düzeltir. Uyarılmış durum A – H / D bağ titreşimlerine sahip olmak için yeterli enerjiye sahip moleküllerin fraksiyonu, oda sıcaklığında veya buna yakın reaksiyonlar için genellikle küçüktür (hidrojene bağlar genellikle 1000 cm−1 veya üstü, yani exp (-senben) = exp (-hνben/kBT) 298 K'da <0.01, 1 – exp (-senben) faktörler). Bu nedenle, hidrojen / döteryum kinetik izotop etkileri için, gözlemlenen değerlere tipik olarak son faktör hakimdir, ZPE (titreşimsel sıfır noktası enerji farklılıklarının üstel bir işlevi), aşağıdaki gibi gösterilebilen, reaktanların titreşim modlarının her biri için sıfır noktası enerji farklılıklarından gelen katkılardan oluşur:[7]
- ,
![{ displaystyle { begin {align} { frac {k _ {{ ce {H}}}} {k _ {{ ce {D}}}}} & cong exp left {- { frac {1} {2}} sol [ sum limits _ {i = 1} ^ {3N ^ { ddagger} -7} (u_ {i { ce {H}}} ^ { ddagger} -u_ {i { ce {D}}} ^ { ddagger}) - sum limits _ {i = 1} ^ {3N-6} (u_ {i { ce {H}}} - u_ {i { ce {D}}}) sağ] sağ } & cong exp left [ sum _ {i} ^ { mathrm {(tepki)}} { frac {1} {2 }} Delta u_ {i} - sum _ {i} ^ { mathrm {(TS)}} { frac {1} {2}} Delta u_ {i} ^ { ddagger} sağ] son {hizalı}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/20d669d624e10fcb89d116442c5543998ea4fbf8)
nerede tanımlıyoruz
- ve .


İkinci ifadenin üssündeki toplamlar, tepkimeye giren temel durum ve geçiş durumunun tüm titreşim modları üzerinden geçiyor olarak yorumlanabilir. Alternatif olarak, bunlar, reaktan veya geçiş durumuna özgü olan modların üzerinden geçerken veya reaksiyon koordinatı boyunca ilerledikçe titreşim frekansları büyük ölçüde değişen olarak yorumlanabilir. Kalan reaktan çiftleri ve geçiş durumu titreşim modları çok benzer ve , ve iptaller üs içindeki toplamlar hesaplandığında gerçekleşir. Bu nedenle, pratikte döteryum KIE'leri, bu iptal nedeniyle genellikle büyük ölçüde bir avuç anahtar titreşim moduna bağımlıdır ve kalitatif analizler yapar. kH/kD mümkün.[13]


Belirtildiği gibi, özellikle hidrojen / döteryum ikamesi için, çoğu kinetik izotop etkisi aşağıdaki farktan kaynaklanır: sıfır nokta enerjisi (ZPE) reaktanlar ile söz konusu izotopologların geçiş durumu arasındaki fark ve bu fark niteliksel olarak aşağıdaki açıklama ile anlaşılabilir: Born-Oppenheimer yaklaşımı potansiyel enerji yüzeyi her iki izotopik tür için aynıdır. Bununla birlikte, enerjinin kuantum-mekanik bir muamelesi, bu eğriye ayrı titreşim seviyeleri getirir ve bir molekülün mümkün olan en düşük enerji durumu, enerji açısından potansiyel enerji eğrisinin minimumundan biraz daha yüksek olan en düşük titreşim enerji seviyesine karşılık gelir. Sıfır noktası enerjisi olarak adlandırılan bu fark, C-H veya C-D bağ uzunluğunda bir belirsizlik gerektiren Heisenberg belirsizlik ilkesinin bir tezahürüdür. Daha ağır olan (bu durumda döteryumlanmış) tür daha "klasik" davrandığından, titreşimsel enerji seviyeleri klasik potansiyel enerji eğrisine daha yakındır ve daha düşük sıfır noktası enerjisine sahiptir. İki izotopik tür arasındaki sıfır noktası enerji farklılıkları, en azından çoğu durumda, bağ kırılması sırasında bağ kuvveti sabiti düştüğü için geçiş durumunda azalır. Dolayısıyla, döteryumlanmış türlerin düşük sıfır noktası enerjisi, aşağıdaki şekilde gösterildiği gibi, reaksiyonu için daha büyük bir aktivasyon enerjisine dönüşerek normal bir kinetik izotop etkisine yol açar.[15] Bu etki, ilke olarak, 3N−Başlangıç malzemesi için 6 titreşim modu ve 3N‡−Geçiş durumunda 7 titreşim modu (bir mod, reaksiyon koordinatına karşılık gelen mod, geçiş durumunda eksiktir, çünkü bir bağ kırılır ve harekete karşı herhangi bir restoratif kuvvet yoktur). harmonik osilatör en azından düşük enerjili titreşim durumları için titreşimli bir bağ için iyi bir yaklaşımdır. Kuantum mekaniği titreşim sıfır noktası enerjisini şu şekilde verir: . Böylece, ½ faktörünü ve toplamlarını kolayca yorumlayabiliriz Temel durum ve geçiş durumu titreşim modları üzerine terimler, yukarıdaki basitleştirilmiş formülün üssünde. Harmonik bir osilatör için titreşim frekansı, titreşimli sistemin azaltılmış kütlesinin kareköküyle ters orantılıdır:


- ,

nerede kf ... kuvvet sabiti. Dahası, indirgenmiş kütle, sistemin ışık atomunun kütlesi X = H veya D ile yaklaşık olarak belirlenir. mD yaklaşık 2mH,
- .

Homolitik bir C-H / D bağ ayrışması durumunda, geçiş durumu terimi kaybolur ve diğer titreşim modlarını ihmal eder, kH/kD = exp (½Δsenben). Bu nedenle, daha sert ("daha güçlü") bir C – H / D bağı için daha büyük bir izotop etkisi gözlenir. İlgili çoğu reaksiyon için, bir hidrojen atomu iki atom arasında bir geçiş durumu [A ··· H ··· B] ile aktarılır.‡ ve geçiş durumundaki titreşim modlarının hesaba katılması gerekir. Yine de, daha yüksek titreşim frekansına sahip bir bağın bölünmesinin daha büyük bir izotop etkisi vereceği genel olarak doğrudur.

Tünel oluşturmayan döteryum KIE için mümkün olan maksimum değeri hesaplamak için, tipik bir karbon-hidrojen bağının (3000 cm) gerilme titreşimleri arasındaki sıfır noktası enerji farkının olduğu durumu dikkate alıyoruz.−1) ve karbon-döteryum bağı (2200 cm−1) geçiş durumunda kaybolur ((1/2) enerji farkı (3000-2200 cm−1) = 400 cm−1, veya yaklaşık 1.15 kcal / mol), geçiş durumunda sıfır noktası enerji farkından herhangi bir telafi olmadan (örneğin, geçiş durumuna özgü simetrik A ··· H ··· B gerilmesinden). Yukarıda verilen basitleştirilmiş formül, bir maksimum kH/kD 6.9 olarak. İki bükülme titreşiminin tamamen ortadan kalkması da dahil edilmişse, kH/kD 15-20 kadar büyük değerler tahmin edilebilir. Bununla birlikte, geçiş durumunda bükülme frekanslarının kaybolması pek olası değildir ve sadece birkaç durum vardır. kH/kD değerler oda sıcaklığına yakın 7-8'i aşıyor. Dahası, tünellemenin bu tür değerleri aştıklarında önemli bir faktör olduğu sıklıkla görülür. Bir değer kH/kD 298 K civarında gerçekleşen reaksiyonlar için yarı klasik birincil kinetik izotop etkisi (tünelleme yok) için ~ 10'un maksimal olduğu düşünülmektedir. kH/kD sıcaklığa bağımlıdır, bu nedenle daha düşük sıcaklıklarda daha büyük izotop etkileri mümkündür).[16] H-transferinin geçiş durumunun doğasına bağlı olarak (simetrik - "erken" veya "geç" ve doğrusal - bükülmüş), birincil döteryum izotop etkisinin bu maksimuma yaklaşma derecesi değişir. Tarafından geliştirilen bir model Westheimer simetrik olduğunu tahmin etti (termonötr, Hammond Postülatı ), doğrusal geçiş durumları en büyük izotop etkilerine sahipken, "erken" veya "geç" (sırasıyla ekzotermik veya endotermik reaksiyonlar için) veya doğrusal olmayan (ör. döngüsel) geçiş durumları daha küçük etkiler sergiler. Bu tahminler o zamandan beri kapsamlı deneysel destek aldı.[17]
İkincil döteryum izotop etkileri için, Streitwieser reaktan temel durumdan geçiş durumuna bükülme modlarının zayıflamasının (veya ters izotop etkisi durumunda güçlendirilmesinin), gözlemlenen izotop etkilerinden büyük ölçüde sorumlu olduğunu öne sürmüştür. Bu değişiklikler, H / D'ye bağlı karbon sp'den yeniden hibridizasyona uğradığında sterik ortamdaki bir değişikliğe atfedilir.3 sp için2 veya tam tersi (bir α ikincil kinetik izotop etkisi) veya bir karbon atomu uzakta bir karbokasyonun üretildiği durumlarda hiper konjugasyon nedeniyle bağ zayıflaması (bir hyper ikincil kinetik izotop etkisi). Bu izotop etkilerinin teorik olarak maksimum kH/kD = 20.5 ≈ 1.4. Α konumunda ikincil kinetik izotop etkisi için, sp'den yeniden hibridizasyon3 sp için2 sp'den yeniden hibridizasyon yaparken normal bir izotop etkisi üretir2 sp için3 teorik minimum değer ile ters izotop etkisi ile sonuçlanır. kH/kD = 2-0.5 ≈ 0.7. Uygulamada, kH/kD ~ 1.1-1.2 ve kH/ kD ~ 0.8-0.9, α ikincil kinetik izotop etkileri için tipikken kH/kD ~ 1.15-1.3, β ikincil kinetik izotop etkisi için tipiktir. Birkaç izotopik olarak ikame edilmiş β-hidrojen atomu içeren reaktanlar için, gözlemlenen izotop etkisi genellikle β konumunda birlikte hareket eden birkaç H / D'nin sonucudur. Bu durumlarda, izotopik olarak etiketlenmiş her atomun etkisi çarpımsaldır ve kH/kD > 2 nadir değildir.[18]
Döteryum ve trityum kinetik izotop etkileri ile ilgili aşağıdaki basit ifadeler; Swain denklemi (veya Swain-Schaad-Stivers denklemleri), bazı basitleştirmeler kullanılarak yukarıda verilen genel ifadeden türetilebilir:[8][19]
- ;

yani
- .

Bu ifadelerin türetilmesinde, indirgenmiş kütlelerin yaklaşık olarak hidrojen, döteryum veya trityum kütlelerine eşit olduğuna dair makul tahmin kullanılmıştır. Ek olarak, titreşim hareketinin bir harmonik osilatör tarafından yaklaştırıldığı varsayıldı, böylece (X = H, D veya T). Alt simge "s", kuantum tünellemeyi göz ardı eden bu" yarı klasik "kinetik izotop etkilerine atıfta bulunur. Tünelleme katkıları, bir düzeltme faktörü olarak ayrı ayrı ele alınmalıdır.

Hidrojen dışındaki elementleri içeren izotop etkileri için, bu basitleştirmelerin çoğu geçerli değildir ve izotop etkisinin büyüklüğü, ihmal edilen faktörlerin bir kısmına veya tümüne büyük ölçüde bağlı olabilir. Bu nedenle, hidrojen dışındaki elementler için kinetik izotop etkilerinin rasyonelleştirilmesi veya yorumlanması genellikle çok daha zordur. Çoğu durumda ve özellikle hidrojen transfer reaksiyonları için, tünellemeden kaynaklanan kinetik izotop etkilerine katkılar önemlidir (aşağıya bakınız).
Tünel açma
Bazı durumlarda, muhtemelen buna bağlı olarak, daha hafif izotop için ek bir hız artışı görülür. kuantum mekanik tünelleme. Bu tipik olarak sadece hidrojen atomlarına bağ içeren reaksiyonlarda gözlemlenir.Tünelleme, bir molekülün üzerinden değil potansiyel bir enerji bariyerinden geçtiğinde meydana gelir.[20][21] Yasaların izin vermemesine rağmen Klasik mekanik parçacıklar, kuantum mekaniğinde klasik olarak yasaklanmış uzay bölgelerinden geçebilir. dalga-parçacık ikiliği.[22]
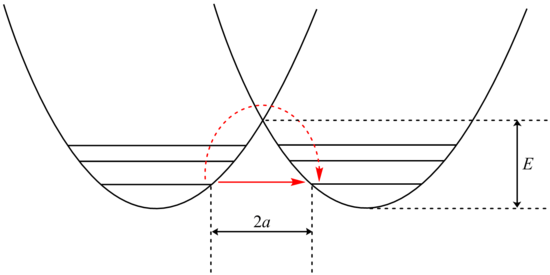
Tünelleme analizi, Bell'in Arrhenius denklemi, bir tünelleme faktörünün eklenmesini içeren Q:

A, Arrhenius parametresidir, E, bariyer yüksekliğidir ve

nerede ve


Muayenesi β terim, parçacığın kütlesine üstel bağımlılığı gösterir. Sonuç olarak, tünel açma, hidrojen gibi daha hafif bir parçacık için çok daha olasıdır. Tünel oluşturan bir protonun kütlesini onun yerine geçerek ikiye katlamak yeterlidir. döteryum izotop, bu tür reaksiyonların oranını büyük ölçüde azaltır. Sonuç olarak, sıfır noktası enerjilerindeki farklılıklarla açıklanamayan çok büyük kinetik izotop etkileri gözlenir.

ek olarak β terim, bariyer genişliğine doğrusal olarak bağlıdır, 2a. Kütle ile olduğu gibi, tünel açma küçük bariyer genişlikleri için en iyisidir. Donör ve alıcı atom arasındaki protonların optimum tünelleme mesafeleri 0,4 Å'dur.[24]
Tünel açma dalga mekaniği yasalarına bağlı bir kuantum mekanik etkidir, değil kinetik. Bu nedenle, tünel açma, en küçük kinetik enerji engellerinin bile aşılamayacağı, ancak tünel açılabildiği düşük sıcaklıklarda daha önemli hale gelme eğilimindedir.[20]
Peter S. Zuev vd. 1-metilsiklobutilflorokarbenin halka genişlemesi için hız sabitlerinin 4.0 x 10 olduğu bildirildi−6/ s nitrojen ve 4.0 x 10−58 kelvin'de argon içinde / s. 8 kelvin'de, reaksiyonun reaktantın tek bir kuantum durumu yoluyla ilerleyeceğini hesapladılar, böylece bildirilen hız sabiti sıcaklıktan bağımsızdır ve hıza tünelleme katkısı geçiş durumu üzerindeki geçişin katkısından 152 derece daha büyüktür. enerji engeli.[25]
Bu nedenle, geleneksel kimyasal reaksiyonların sıcaklık düştükçe çarpıcı bir şekilde yavaşlama eğiliminde olmasına rağmen, tünel oluşturma reaksiyonları nadiren hiç değişir. Bir aktivasyon bariyerinden tünel açan parçacıklar, bir ara tür, reaktan veya ürünün dalga fonksiyonunun, bir reaksiyonun enerji yüzeyi boyunca belirli bir çukurun enerji kuyusuyla sınırlı olmadığı, ancak "dışarı sızabileceği" gerçeğinin doğrudan bir sonucudur. bir sonraki enerji minimumuna. Bunun ışığında tünel açma meli sıcaklıktan bağımsız olun.[20][3]
363-463 K sıcaklık aralığı üzerindeki hidrojen atomları tarafından gaz halindeki n-alkanlardan ve sikloalkanlardan hidrojen soyutlaması için, H / D kinetik izotop etkisi verileri küçük ön üstel faktör oranlar BirH/BirD 0,43 ila 0,54 arasında değişen ve 9,0 ila 9,7 kJ / mol arasında büyük aktivasyon enerjisi farklılıkları. Argümanlarını temel alarak geçiş durumu teorisi, küçük Bir Büyük aktivasyon enerjisi farklılıklarıyla ilişkili faktör oranları (genellikle C-H (D) bağları için yaklaşık 4,5 kJ / mol) tünel oluşturma için güçlü kanıtlar sağlamıştır. Bu tartışmanın amacı için, önemli olan, Bir kullandıkları çeşitli parafinler için faktör oranı, sıcaklık aralığı boyunca yaklaşık olarak sabitti.[26]
Tünel açmanın tamamen sıcaklıktan bağımsız olmadığı gözlemi, belirli bir türe ait tüm moleküllerin değişen sıcaklıklarda titreşimsel temel durumlarını işgal etmedikleri gerçeğiyle açıklanabilir. Potansiyel bir enerji kuyusuna termal enerji eklemek, temel durum dışındaki daha yüksek titreşim seviyelerinin doldurulmasına neden olabilir. Geleneksel kinetik olarak tahrik edilen bir reaksiyon için, bu uyarmanın hız üzerinde sadece küçük bir etkisi olacaktır. Bununla birlikte, bir tünelleme reaksiyonu için, arasındaki fark sıfır noktası enerjisi ve ilk titreşimsel enerji seviyesi çok büyük olabilir. Tünel açma düzeltme terimi Q bariyer genişliğine doğrusal olarak bağlıdır ve bu genişlik, sayı arttıkça önemli ölçüde azalır. titreşim modları üzerinde Mors potansiyeli artırmak. Bariyer genişliğinin azalması tünel açma hızı üzerinde o kadar büyük bir etkiye sahip olabilir ki, küçük bir uyarılmış titreşim durumları popülasyonu bile bu sürece hakim olabilir.[20][3]H veya D ile bir reaksiyonun KIE'sine tünel oluşturmanın dahil olup olmadığını belirlemek için birkaç kriter dikkate alınır:
- Δ (EaH-EaD)> Δ (ZPEH-ZPED) (Ea= aktivasyon enerjisi; ZPE = sıfır noktası enerjisi)
- Reaksiyon hala daha düşük sıcaklıklarda devam etmektedir.
- Arrhenius üstel faktörler BirD/BirH 1'e eşit değildir.
- Büyük bir negatif entropi aktivasyon.
- Reaktiflerin ve ürünlerin geometrileri genellikle çok benzerdir.[20]
Ayrıca izotopların H, D ve T'yi içerdiği reaksiyonlar için, hız sabitlerini karşılaştıran Swain-Schaad ilişkilerinde tünelleme kriteri birk) H, D veya T'nin değiştirildiği reaksiyonların:
- kH/kT=(kD/kT)X ve kH/kT=(kH/kD)Y
Organik reaksiyonlarda, bu proton tünelleme etkisi, aşağıdaki gibi reaksiyonlarda gözlenmiştir. protonsuzlaşma ve iyotlama nitropropan engellenmiş piridin temel[27] 25 ° C'de rapor edilen KIE 25 ile:

ve içinde 1,5-sigmatropik hidrojen kayması[28] yüksek sıcaklıklarda elde edilen deneysel değerlerin daha düşük sıcaklıklara çıkarılmasının zor olduğu gözlemlenmesine rağmen:[29][30]

Proton veya hidrit iyon transfer reaksiyonlarında enzim katalizinin yüksek verimliliğinin kısmen kuantum mekanik tünelleme etkisine bağlı olabileceği uzun süredir tahmin edilmektedir. Bir enzimin aktif bölgesindeki ortam, verici ve alıcı atomu optimum tünel açma mesafesine yakın konumlandırır, burada amino asit yan zincirleri, elektrostatik ve kovalent olmayan etkileşimlerle verici ve alıcı atomu birbirine daha yakın "zorlayabilir". Bir reaksiyon bölgesi içindeki enzim ve olağandışı hidrofobik ortamın tünel açmayı teşvik eden titreşim sağlaması da mümkündür.[31] Studies on ketosteroid isomerase have provided experimental evidence that the enzyme actually enhances the coupled motion/hydrogen tunneling by comparing primary and secondary kinetic isotope effects of the reaction under enzyme catalyzed and non-enzyme catalyzed conditions.[32]
Many examples exist for proton tunneling in enzyme catalyzed reactions that were discovered by KIE. A well studied example is methylamine dehydrogenase, where large primary KIEs of 5–55 have been observed for the proton transfer step.[33]

Another example of tunneling contribution to proton transfer in enzymatic reactions is the reaction carried out by alkol dehidrojenaz. Competitive KIEs for the hydrogen transfer step at 25 °C resulted in 3.6 and 10.2 for primary and secondary KIEs, respectively.[34]
Transient kinetic isotope effect
Isotopic effect expressed with the equations given above only refer to reactions that can be described with birinci dereceden kinetik. In all instances in which this is not possible, transient kinetic isotope effects should be taken into account using the GEBIK and GEBIF equations.[35]
Deneyler
Simmons and Hartwig refer to the following three cases as the main types of kinetic isotope effect experiments involving C-H bond functionalization:[5]
- A) KIE determined from absolute rates of two parallel reactions

In this experiment, the rate constants for the normal substrate and its isotopically labeled analogue are determined independently, and the KIE is obtained as a ratio of the two. The accuracy of the measured KIE is severely limited by the accuracy with which each of these rate constants can be measured. Furthermore, reproducing the exact conditions in the two parallel reactions can be very challenging. Nevertheless, a measurement of a large kinetic isotope effect through direct comparison of rate constants is indicative that C-H bond cleavage occurs at the rate-determining step. (A smaller value could indicate an isotope effect due to a pre-equilibrium, so that the C-H bond cleavage occurs somewhere before the rate-determining step.)
- B) KIE determined from an intermolecular competition
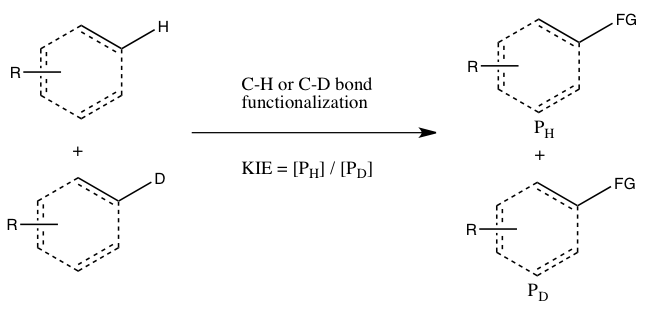
In this type of experiment, the same substrates that are used in Experiment A are employed, but they are allowed in to react in the same container, instead of two separate containers. The kinetic isotope effect from this experiment is determined by the relative amount of products formed from C-H versus C-D functionalization (or it can be inferred from the relative amounts of unreacted starting materials). It is necessary to quench the reaction before it goes to completion to observe the kinetic isotope effect (see the Evaluation section below). Generally, the reaction is halted at low conversion (~5 to 10% conversion) or a large excess (> 5 equiv.) of the isotopic mixture is used. This experiment type ensures that both C-H and C-D bond functionalizations occur under exactly the same conditions, and the ratio of products from C-H and C-D bond functionalizations can be measured with much greater precision than the rate constants in Experiment A. Moreover, only a single measurement of product concentrations from a single sample is required. However, an observed kinetic isotope effect from this experiment is more difficult to interpret, since it may either mean that C-H bond cleavage occurs during the rate-determining step or at a product-determining step ensuing the rate-determining step. The absence of a kinetic isotope effect, at least according to Simmons and Hartwig, is nonetheless indicative of the C-H bond cleavage not occurring during the rate-determining step.
- C) KIE determined from an intramolecular competition
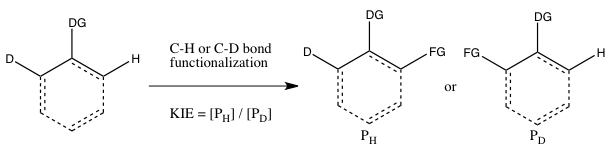
This type of experiment is analogous to Experiment B, except this time there is an intramolecular competition for the C-H or C-D bond functionalization. In most cases, the substrate possesses a directing group (DG) between the C-H and C-D bonds. Calculation of the kinetic isotope effect from this experiment and its interpretation follow the same considerations as that of Experiment B. However, the results of Experiments B and C will differ if the irreversible binding of the isotope-containing substrate takes place in Experiment B önceki to the cleavage of the C-H or C-D bond. In such a scenario, an isotope effect may be observed in Experiment C (where choice of the isotope can take place even after substrate binding) but not in Experiment B (since the choice of whether C-H or C-D bond cleaves is already made as soon as the substrate binds irreversibly). In contrast to Experiment B, the reaction does not need to be halted at low consumption of isotopic starting material to obtain an accurate kH/kD, since the ratio of H and D in the starting material is 1:1, regardless of the extent of conversion.
One non-C-H activation example of different isotope effects being observed in the case of intermolecular (Experiment B) and intramolecular (Experiment C) competition is the photolysis of diphenyldiazomethane in the presence of t-butylamine. To explain this result, the formation of diphenylcarbene, followed by irreversible nucleophilic attack by t-butylamine was proposed. Because there is little isotopic difference in the rate of nucleophilic attack, the intermolecular experiment resulted in a KIE close to 1. In the intramolecular case, however, the product ratio is determined by the proton transfer that occurs after the nucleophilic attack, a process for which there is a substantial KIE of 2.6.[36]

Thus, Experiments A, B, and C will give results of differing levels of precision and require different experimental setup and ways of analyzing data. As a result, the feasibility of each type of experiment will depend on the kinetic and stoichiometric profile of the reaction, as well as the physical characteristics of the reaction mixture (e.g., homogeneous vs. heterogeneous). Moreover, as noted in the paragraph above, the experiments provide kinetic isotope effect data for different steps of a multi-step reaction, depending on the relative locations of the rate-limiting step, product-determining steps, and/or C-H/D cleavage step.
The hypothetical examples below illustrate common scenarios. Consider the following reaction coordinate diagram. For a reaction with this profile, all three experiments (A, B, and C) will yield a significant primary kinetic isotope effect:

On the other hand, if a reaction follows the following energy profile, in which the C-H or C-D bond cleavage is irreversible but occurs after the rate-determining step (RDS), no significant kinetic isotope effect will be observed with Experiment A, since the overall rate is not affected by the isotopic substitution. Nevertheless, the irreversible C-H bond cleavage step will give a primary kinetic isotope effect with the other two experiments, since the second step would still affect the product distribution. Therefore, with Experiments B and C, it is possible to observe the kinetic isotope effect even if C-H or C-D bond cleavage occurs not in the rate-determining step, but in the product-determining step.

A large part of the kinetic isotope effect arises from vibrational zero-point energy differences between the reactant ground state and the transition state that vary between the reactant and its isotopically substituted analogue. While it is possible to carry involved calculations of kinetic isotope effects using computational chemistry, much of the work done is of simpler order that involves the investigation of whether particular isotopic substitutions produce a detectable kinetic isotope effect or not. Vibrational changes from isotopic substitution at atoms away from the site where the reaction occurs tend to cancel between the reactant and the transition state. Therefore, the presence of a kinetic isotope effect indicates that the isotopically labeled atom is at or very near the reaction site.
The absence of an isotope effect is more difficult to interpret: It may mean that the isotopically labeled atom is away from the reaction site, but it may also mean that there are certain compensating effects that lead to the lack of an observable kinetic isotope effect. For example, the differences between the reactant and the transition state zero-point energies may be identical between the normal reactant and its isotopically labeled version. Alternatively, it may mean that the isotopic substitution is at the reaction site, but vibrational changes associated with bonds to this atom occur after the rate-determining step. Such a case is illustrated in the following example, in which ABCD represents the atomic skeleton of a molecule.

Assuming steady state conditions for the intermediate ABC, the overall rate of reaction is the following:
![{ frac {d [A]} {dt}} = { frac {k_ {1} k_ {3} [ABCD]} {k_ {2} [D] + k_ {3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f71c52221df559ae3b305086cf125e2cfbfa62c4)
If the first step is rate-determining, this equation reduces to:
![{ frac {d [A]} {dt}} = k_ {1} [ABCD]](https://wikimedia.org/api/rest_v1/media/math/render/svg/6cfaea2beb4c13e320a49b4f66ede1c5a85d0fba)
Or if the second step is rate-determining, the equation reduces to:
![{ frac {d [A]} {dt}} = { frac {k_ {1} k_ {3} [ABCD]} {k_ {2} [D]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ea50a8acf1a1307f4d9c2b4605f62578e2f79771)
In most cases, isotopic substitution at A, especially if it is a heavy atom, will not alter k1 veya k2, but it will most probably alter k3. Hence, if the first step is rate-determining, there will not be an observable kinetic isotope effect in the overall reaction with isotopic labeling of A, but there will be one if the second step is rate-determining. For intermediate cases where both steps have comparable rates, the magnitude of the kinetic isotope effect will depend on the ratio of k3 ve k2.
Isotopic substitution of D will alter k1 ve k2 while not affecting k3. The kinetic isotope effect will always be observable with this substitution since k1 appears in the simplified rate expression regardless of which step is rate-determining, but it will be less pronounced if the second step is rate-determining due to some cancellation between the isotope effects on k1 ve k2. This outcome is related to the fact that equilibrium isotope effects are usually smaller than kinetic isotope effects.
Isotopic substitution of B will clearly alter k3, but it may also alter k1 to a lesser extent if the B-C bond vibrations are affected in the transition state of the first step. There may thus be a small isotope effect even if the first step is rate-determining.
This hypothetical consideration reveals how observing kinetic isotope effects may be used to investigate reaction mechanisms. The existence of a kinetic isotope effect is indicative of a change to the vibrational force constant of a bond associated with the isotopically labeled atom at or before the rate-controlling step. Intricate calculations may be used to learn a great amount of detail about the transition state from observed kinetic isotope effects. More commonly, though, the mere qualitative knowledge that a bond associated with the isotopically labeled atom is altered in a certain way can be very useful.[37]Evaluation of rate constant ratios from intermolecular competition reactions
In competition reactions, the kinetic isotope effect is calculated from isotopic product or remaining reactant ratios after the reaction, but these ratios depend strongly on the extent of completion of the reaction. Most commonly, the isotopic substrate will consist of molecules labeled in a specific position and their unlabeled, ordinary counterparts.[8] It is also possible in case of 13C kinetic isotope effects, as well as similar cases, to simply rely on the natural abundance of the isotopic carbon for the kinetic isotope effect experiments, eliminating the need for isotopic labeling.[38] The two isotopic substrates will react through the same mechanism, but at different rates. The ratio between the amounts of the two species in the reactants and the products will thus change gradually over the course of the reaction, and this gradual change can be treated in the following manner:[8]Assume that two isotopic molecules, A1 ve A2, undergo irreversible competition reactions in the following manner:
![{ displaystyle { begin {align} { ce {{A1} + {B} + {C} + cdots}} & { ce {-> [k_ {1}] P1}} { ce {{A2} + {B} + {C} + cdots}} & { ce {-> [k_ {2}] P2}} end {hizalı}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8a3e1c66994bddc0e6b39e89f23e4e0ec7a47b5c)
The kinetic isotope effect for this scenario is found to be:

Where F1 ve F2 refer to the fraction of conversions for the isotopic species A1 ve A2, sırasıyla.
In this treatment, all other reactants are assumed to be non-isotopic. Assuming further that the reaction is of first order with respect to the isotopic substrate A, the following general rate expression for both these reactions can be written:
![{ displaystyle { text {oran}} = {- d [{ ce {A}} _ {n}] over dt} = k_ {n} times [{ ce {A}} _ {n} ] times f ([{ ce {B}}], [{ ce {C}}], cdots) { text {nerede}} n = 1 { text {veya}} 2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/26ca6ed80abd2295998fb23de5de165477f2848e)
Since f([B],[C],…) does not depend on the isotopic composition of A, it can be solved for in both rate expressions with A1 ve A2, and the two can be equated to derive the following relations:
![{ displaystyle {1 over k_ {1}} times { ce {{ mathit {d}} [A1] over [A1]}} = {1 over k_ {2}} times { ce {{ mathit {d}} [A2] over [A2]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ec95ce0a836f8db29e8a86da2d3a2390214f462c)
![{ displaystyle {1 over k_ {1}} times int limits _ { ce {[A1] ^ {0}}} ^ { ce {[A1]}} {d [{ ce {A }} '_ {1}] over [{ ce {A}}' _ {1}]} = {1 over k_ {2}} times int limits _ { ce {[A2] ^ {0}}} ^ { ce {[A2]}} {d [{ ce {A}} '_ {2}] over [{ ce {A}}' _ {2}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8b3f5c1311b00398d60afa93ae27d58ac423fd32)
Where [A1]0 and [A2]0 are the initial concentrations of A1 ve A2, sırasıyla. This leads to the following kinetic isotope effect expression:
![{ displaystyle {k_ {1} over k_ {2}} = { frac { ce { ln ([A1] / [A1] ^ {0})}} { ce { ln ([A2] / [A2] ^ {0})}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a4f8b9f334d8799e9e6fc51d105e53d0148086f1)
Which can also be expressed in terms of fraction amounts of conversion of the two reactions, F1 ve F2, where 1-Fn=[An]/[An]0 for n = 1 or 2, as follows:


As for obtaining the kinetic isotope effects, mixtures of substrates containing stable isotopes may be analyzed using a mass spectrometer, which yields the ratios of the isotopic molecules in the initial substrate (defined here as [A2]0/[A1]0= R0), in the substrate after some conversion ([A2]/[A1]=R), or in the product ([P2]/[P1]=RP). When one of the species, e.g. 2, is a radioactive isotope, its mixture with the other species can also be analyzed by its radioactivity, which is measured in molar activities that are proportional to [A2]0 / ([A1]0+[A2]0) ≈ [A2]0/[A1]0 = R0 in the initial substrate, [A2] / ([A1]+[A2]) ≈ [A2]/[A1] = R in the substrate after some conversion, and [R2] / ([R1]+[R2]) ≈ [R2]/[R1] = RP, so that the same ratios as in the other case can be measured as long as the radioactive isotope is present in tracer amounts. Such ratios may also be determined using NMR spectroscopy.[39][40]
When the substrate composition is followed, the following kinetic isotope effect expression in terms of R0 and R can be derived:
![{ displaystyle { text {KIE}} = { frac {k_ {1}} {k_ {2}}} = { frac { ln (1-F_ {1})} { ln [(1- F_ {1}) R / R_ {0}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9f819291ca2e6641dfaf1a2d31025c40927bdd7a)
Taking the ratio of R and R0 using the previously derived expression for F2, one gets:
![{ displaystyle {R over R_ {0}} = { ce {{ frac {[A2] / [A1]} {[A2] ^ 0 / [A1] ^ 0}}}} = { ce { { frac {[A2] / [A2] ^ 0} {[A1] / [A1] ^ 0}}}} = { frac {1-F_ {2}} {1-F_ {1}}} = (1-F_ {1}) ^ {(k_ {2} / k_ {1}) - 1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/61529d236e7c75ed167c0a1e43f996134b54b908)
Isotopic enrichment of the starting material can be calculated from the dependence of R / R0 açık F1 for various kinetic isotope effects, yielding the following figure. Because of the exponential dependence, even very low kinetic isotope effects lead to large changes in isotopic composition of the starting material at high conversions.

When the products are followed, the kinetic isotope effect can be calculated using the products ratio RP ile birlikte R0 aşağıdaki gibi:
![{k_ {1} over k_ {2}} = { frac { ln (1-F_ {1})} { ln [1- (F_ {1} R_ {P} / R_ {0})] }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/37f721659b133bfa69404286f90a8309fea92944)
Kinetic isotope effect measurement at natural abundance
Kinetic isotope effect measurement at natural abundance is a simple general method for measuring kinetic isotope effects (KIE) for kimyasal reaksiyonlar performed with materials of doğal bolluk. This technique for measuring KIEs overcomes many limitations of previous KIE measurement methods. KIE measurements from isotopically labeled materials require a new synthesis for each isotopically labeled material (a process often prohibitively difficult), a competition reaction, and an analysis.[5] The KIE measurement at doğal bolluk avoids these issues by taking advantage of high precision quantitative techniques (nükleer manyetik rezonans Spektroskopisi, izotop oranı kütle spektrometresi ) to site selectively measure kinetik fraksiyonlama nın-nin izotoplar, in either product or starting material for a given Kimyasal reaksiyon.
Single-pulse NMR
Quantitative single-pulse nükleer manyetik rezonans Spektroskopisi (NMR) is a method amenable for measuring kinetik fraksiyonlama nın-nin izotoplar for natural abundance KIE measurements. Pascal et al. were inspired by studies demonstrating dramatic variations of deuterium within identical compounds from different sources and hypothesized that NMR could be used to measure deuterium kinetic isotope effects at natural abundance.[41][42] Pascal and coworkers tested their hypothesis by studying the ekleme reaksiyonu of dimethyl diazomalonate into sikloheksan. Pascal et al. measured a KIE of 2.2 using 2
H
NMR for materials of natural abundance.[42]

Singleton and coworkers demonstrated the capacity of 13
C
NMR based natural abundance KIE measurements for studying the mechanism of the [4 + 2] siklokasyon nın-nin izopren ile maleik anhidrit.[38] Previous studies by Gajewski on isotopically enrich materials observed KIE results that suggested an asynchronous transition state, but were always consistent, within error, for a perfectly synchronous reaksiyon mekanizması.[43]

This work by Singleton et al. established the measurement of multiple 13
C
KIE's within the design of a single experiment. Bunlar 2
H
ve 13
C
KIE measurements determined at natural abundance found the “inside” hydrogens of the diene experience a more pronounced 2
H
KIE than the “outside” hydrogens” and the C1 and C4 experience a significant KIE. These key observations suggest an asynchronous reaksiyon mekanizması için siklokasyon nın-nin izopren ile maleik anhidrit.
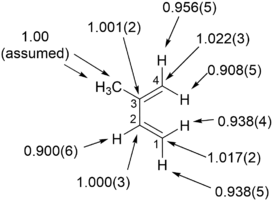
The limitations for determining KIE's at natural abundance using NMR are that the recovered material must have a suitable amount and purity for NMR analysis (the signal of interest should be distinct from other signals), the reaction of interest must be irreversible, and the reaksiyon mekanizması must not change for the duration of the Kimyasal reaksiyon.
Experimental details for using quantitative single pulse NMR to measure kinetic isotope effect at natural abundance as follows: the experiment needs to be performed under quantitative conditions including a relaxation time of 5 T1, measured 90° flip angle, a digital resolution of at least 5 points across a peak, and a signal:noise greater than 250. The raw FID is zero-filled to at least 256K points before the Fourier transform. NMR spectra are phased and then treated with a zeroth order baseline correction without any tilt correction. Signal integrations are determined numerically with a minimal tolerance for each integrated signal.[38][açıklama gerekli ]
Organometallic reaction mechanism elucidation examples
Colletto et al. developed a regioselective -arylation of benzo[b]thiophenes at room temperature with aryl iodides as coupling partners and sought to understand the mechanism of this reaction by performing natural abundance kinetic isotope effect measurements via single pulse NMR.[44]




The observation of a primary 13C isotope effect at C3, an inverse 2H isotope effect, a secondary 13C isotope effect at C2, and the lack of an 2H isotope effect at C2 lead Colletto et al. to suggest a Heck-type reaction mechanism for the regioselective -arylation of benzo[b]thiophenes at room temperature with aryl iodides as coupling partners.[44]
Don et al. sought to understand the effects of Lewis asidi additives on the mechanism of enantioselective palladium catalyzed C-N bond activation using natural abundance kinetic isotope effect measurements via single pulse NMR.[45]


Birincil 13C kinetic isotope effect observed in the absence of BPh3 suggests a reaction mechanism with rate limiting cis oxidation into the C–CN bond of the cyanoformamide. The addition of BPh3 causes a relative decrease in the observed 13C kinetic isotope effect which led Frost et al. to suggest a change in the rate limiting step from cis oxidation to coordination of palladium to the cyanoformamide.[45]
DEPT-55 NMR
Although kinetic isotope effect measurements at natural abundance are a powerful tool for understanding reaction mechanisms, the amounts of material required for analysis can make this technique inaccessible for reactions that employ expensive reagents or unstable starting materials. In order to mitigate these limitations, Jacobsen and coworkers developed 1H - 13C polarization transfer as a means to reduce the time and material required for kinetic isotope effect measurements at natural abundance. distortionless enhancement by polarization transfer (DEPT) takes advantage of the larger jiromanyetik oran nın-nin 1H over 13C to theoretically improve measurement sensitivity by a factor of 4 or decrease experiment time by a factor of 16. This method for natural abundance kinetic isotope measurement is favorable for analysis for reactions containing unstable starting materials, and catalysts or products that are relatively costly.[46]
Jacobsen and coworkers identified the thiourea-catalyzed glycosylation of galactose as a reaction that met both of the aforementioned criteria (expensive materials and unstable substrates) and was a reaction with a poorly understood mechanism.[47] Glycosylation is a special case of nucleophilic substitution that lacks clear definition between SN1 ve SN2 mechanistic character. The presence of the oxygen adjacent to the site of displacement (i.e., C1) can stabilize positive charge. This charge stabilization can cause any potential concerted pathway to become asynchronous and approaches intermediates with oxocarbenium character of the SN1 mechanism for glycosylation.


Jacobsen and coworkers observed small normal KIE's at C1, C2, and C5 which suggests significant oxocarbenium character in the transition state and an asynchronous reaction mechanism with a large degree of charge separation.
Isotope-ratio mass spectrometry
Yüksek hassasiyet izotop oranı kütle spektrometresi (IRMS) is another method for measuring kinetik fraksiyonlama nın-nin izotoplar for natural abundance KIE measurements. Widlanski and coworkers demonstrated 34
S
KIE at natural abundance measurements for the hidroliz nın-nin sülfat monoesters. Their observation of a large KIE suggests S-O bond cleavage is rate controlling and likely rules out an associate reaksiyon mekanizması.[48]

The major limitation for determining KIE's at natural abundance using IRMS is the required site selective degradation without isotopic fractionation into an analyzable small molecule, a non-trivial task.[38]
Durum çalışmaları
Primary hydrogen isotope effects
Primary hydrogen kinetic isotope effects refer to cases in which a bond to the isotopically labeled hydrogen is formed or broken at a rate- and/or product-determining step of a reaction.[5] These are the most commonly measured kinetic isotope effects, and much of the previously covered theory refers to primary kinetic isotope effects.When there is adequate evidence that transfer of the labeled hydrogen occurs in the rate-determining step of a reaction, if a fairly large kinetic isotope effect is observed, e.g. kH/kD of at least 5-6 or kH/kT about 10–13 at room temperature, it is quite likely that the hydrogen transfer is linear and that the hydrogen is fairly symmetrically located in the transition state. It is usually not possible to make comments about tunneling contributions to the observed isotope effect unless the effect is very large. If the primary kinetic isotope effect is not as large, it is generally considered to be indicative of a significant contribution from heavy-atom motion to the reaction coordinate, although it may also mean that hydrogen transfer follows a nonlinear pathway.[8]
Secondary hydrogen isotope effects
The secondary hydrogen isotope effects or secondary kinetic isotope effect (SKIE) arises in cases where the isotopic substitution is remote from the bond being broken. The remote atom, nonetheless, influences the internal vibrations of the system that via changes in the zero point energy (ZPE) affect the rates of chemical reactions.[49] Such effects are expressed as ratios of rate for the light isotope to that of the heavy isotope and can be "normal" (ratio is greater than or equal to 1) or "inverse" (ratio is less than 1) effects.[50] SKIE are defined as α, β (etc.) secondary isotope effects where such prefixes refer to the position of the isotopic substitution relative to the reaction center (see alpha and beta carbon ).[51] The prefix α refers to the isotope associated with the reaction center while the prefix β refers to the isotope associated with an atom neighboring the reaction center and so on.
In physical organic chemistry, SKIE is discussed in terms of elektronik efektler such as induction, bond hybridization, or hiperkonjugasyon.[52] These properties are determined by electron distribution, and depend upon vibrationally averaged bond length and angles that are not greatly affected by isotopic substitution. Thus, the use of the term "electronic isotope effect" while legitimate is discouraged from use as it can be misinterpreted to suggest that the isotope effect is electronic in nature rather than vibrational.[51]
SKIE's can be explained in terms of changes in orbital hybridization. When the hybridization of a carbon atom changes from sp3 sp için2, a number of vibrational modes (stretches, in-plane and out-of-plane bending) are affected. The in-plane and out-of-plane bending in an sp3 hybridized carbon are similar in frequency due to the symmetry of an sp3 hybridized carbon. In an sp2 hybridized carbon the in-plane bend is much stiffer than the out-of-plane bending resulting in a large difference in the frequency, the ZPE and thus the SKIE (which exists when there is a difference in the ZPE of the reactant and transition state).[20]The theoretical maximum change caused by the bending frequency difference has been calculated as 1.4.[20]
When carbon undergoes a reaction that changes its hybridization from sp3 sp için2, the out of plane bending force constant at the transition state is weaker as it is developing sp2 character and a "normal" SKIE is observed with typical values of 1.1 to 1.2.[20] Conversely, when carbon's hybridization changes from sp2 sp için3, the out of plane bending force constants at the transition state increase and an inverse SKIE is observed with typical values of 0.8 to 0.9.[20]
More generally the SKIE for reversible reactions can be "normal" one way and "inverse" the other if bonding in the transition state is midway in stiffness between substrate and product, or they can be "normal" both ways if bonding is weaker in the transition state, or "inverse" both ways if bonding is stronger in the transition state than in either reactant.[50]
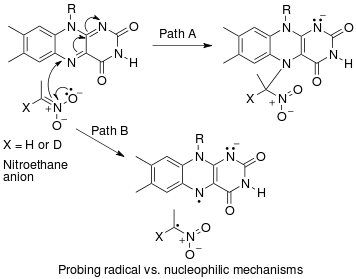
An example of an "inverse" α secondary kinetic isotope effect can be seen in the work of Fitzpatrick and Kurtz who used such an effect to distinguish between two proposed pathways for the reaction of d-amino acid oxidase ile nitroalkane anyonlar.[53] Path A involved a nucleophilic attack on the coenzyme HEVES, while path B involves a free-radical intermediate. As path A results in the intermediate carbon changing hybridization from sp2 sp için3 an "inverse" a SKIE is expected. If path B occurs then no SKIE should be observed as the free radical intermediate does not change hybridization. An SKIE of 0.84 was observed and Path A verified as shown in the scheme below.
Another example of a SKIE is the oxidation of benzyl alcohols by dimethyldioxirane where three transition states for different mechanisms were proposed. Again, by considering how and if the hydrogen atoms were involved in each, researchers predicted whether or not they would expect an effect of isotopic substitution of them. Then, analysis of the experimental data for the reaction allowed them to choose which pathway was most likely based on the observed isotope effect.[54]
Metilen hidrojenlerinden ikincil hidrojen izotop etkileri de 1,5-heksadiende Cope yeniden düzenlemesinin uyumlu bir bağ yeniden düzenleme yolunu izlediğini ve alternatif olarak önerilen allil radikali veya 1,4-diyl yollarından birini olmadığını göstermek için kullanıldı. aşağıdaki şemada sunulmuştur.[55]

1,5-heksadienin Cope yeniden düzenlenmesi için alternatif mekanizmalar: (yukarıdan aşağıya), allil radikali, senkronize uyumlu ve 1,4-dyil yolları. Baskın yolun, aromatik bir ara maddeye karşılık gelen altı delokalize π elektrona sahip olan ortadaki yol olduğu bulunmuştur.[55]
Sterik izotop etkileri
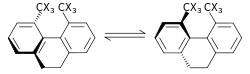 |

Sterik izotop etkisi, bağ kırılması veya oluşumunu içermeyen bir SKIE'dir. Bu etki, farklı titreşim genliklerine atfedilir. izotopologlar.[56] Böyle bir etkinin bir örneği, rasemizasyon 9,10-dihidro-4,5-dimetilfenantren ile karıştırılmıştır.[57] C-H (karbon-hidrojen), C-D (karbon-döteryum) bağlarındaki hidrojene kıyasla döteryum için daha küçük titreşim genliği, daha küçük bir van der Waals yarıçapı veya etkili boyutla sonuçlanırken, ZPE'de iki. Biri diğerini içeren daha büyük bir etkili molekül kütlesi olduğunda, bu, hız sabiti üzerindeki sterik bir etki ile ortaya çıkabilir. Yukarıdaki örnek için döteryum, hidrojen izotopologundan daha hızlı racemizleşir ve sterik bir izotop etkisi ile sonuçlanır. Sterik izotop etkisi için bir model Bartell tarafından geliştirilmiştir.[58] Yukarıda gösterilen rasemizasyon işleminde olduğu gibi, dönüşümler şiddetli sterik yüke sahip bir geçiş durumundan geçmedikçe, sterik bir izotop etkisi genellikle küçüktür.
Sterik izotop etkisinin başka bir örneği, rotaksanların kayma reaksiyonudur. Döteryum izotopu, daha küçük etkili boyutu nedeniyle, tıpaların makrosaykıl boyunca daha kolay geçişine izin vererek döteryumlanmışlar için daha hızlı kayma oranları ile sonuçlanır. rotaksanlar.[59]
Ters kinetik izotop etkileri
Döteryumlanmış türlerin reaksiyona girdiği reaksiyonlar bilinmektedir Daha hızlı kısırlaştırılmamış analoğa göre ve bu durumların ters kinetik izotop etkileri (IKIE) sergilediği söyleniyor. IKIE'ler genellikle indirgeyici eliminasyon alkil metal hidrürler, ör. (Ben mi2NCH2CH2NMe2 ) PtMe (H). Bu gibi durumlarda geçiş durumunda C-D bağı, bir agostik türleri, C – H bağına göre oldukça stabildir.[kaynak belirtilmeli ]
Genel hız sabiti bir şeye bağlıysa, çok adımlı bir reaksiyonda ters etki de meydana gelebilir. ön denge öncesinde oran belirleme adımı tersi olan denge izotop etkisi. Örneğin, oranları asit katalizli D'deki reaksiyonlar için reaksiyonlar genellikle 2-3 kat daha büyüktür2O, D tarafından katalize edildi3Ö+ H'deki benzer reaksiyonlara göre2O, H tarafından katalize edildi3Ö+[4]:433 Bu bir mekanizma için açıklanabilir özgül hidrojen iyon katalizi bir reaktan R, H3Ö+ (veya D3Ö+).
- H3Ö+ + R RH+ + H2Ö
- RH+ + H2O → H3Ö+ + P
Bu durumda ürünlerin oluşum hızı d [P] / dt = k2[RH+] = k2K1[H3Ö+] [R] = kgözlem[H3Ö+] [R]. İlk adımda, H3Ö+ genellikle bağıl nemden daha güçlü bir asittir+. Deuterasyon, dengeyi daha güçlü bir şekilde bağlı asit türleri RD'ye kaydırır.+ sıfır noktası titreşim enerjisi üzerindeki döteryumun etkisinin daha büyük olduğu, böylece döteryumlanmış denge sabiti K1G K'den büyük1H. İlk adımdaki bu denge izotop etkisi, genellikle ikinci adımdaki kinetik izotop etkisinden daha ağır basar, böylece görünür bir ters izotop etkisi vardır ve gözlenen genel hız sabiti kgözlem = k2K1 azalır.[4]:433
Çözücü hidrojen kinetik izotop etkileri
Çözücü izotop etkilerinin ölçülebilir olması için, çözücünün sonlu bir fraksiyonunun geri kalanından farklı bir izotopik bileşime sahip olması gerekir. Bu nedenle, daha az yaygın olan izotopik türlerin büyük miktarları mevcut olmalıdır, bu da gözlemlenebilir çözücü izotop etkilerini hidrojeni içeren izotopik ikamelerle sınırlandırır. Saptanabilir kinetik izotop etkileri, yalnızca çözücüler çözücü ile hidrojeni değiştirdiğinde veya reaksiyon sahası yakınında belirli bir çözücü-çözücü etkileşimi olduğunda ortaya çıkar. Bu tür olayların her ikisi de hidrojenin değiştirilebilir olduğu protik çözücüler için yaygındır ve polar moleküller ile dipol-dipol etkileşimleri veya hidrojen bağları oluşturabilirler.[8]
Karbon-13 izotop etkileri
Çoğu organik reaksiyon, bir karbona bağların kopması ve yapılmasını içerir; bu nedenle saptanabilir karbon izotop etkilerinin beklenmesi mantıklıdır. Ne zaman 13Etiket olarak C kullanılır, izotopun kütlesindeki değişim sadece ~% 8'dir, bu da gözlemlenebilir kinetik izotop etkilerini hidrojen izotop etkileriyle gözlemlenenden çok daha küçük değerlerle sınırlar.
Varyasyonları telafi etme 13C doğal bolluk
Çoğu zaman, karbonun doğal bolluğuna bağlı olan bir çalışmadaki en büyük hata kaynağı, doğal karbon miktarındaki küçük farklılıklardır. 13C bolluğunun kendisi. Bu tür varyasyonlar, reaksiyonda kullanılan başlangıç malzemelerinin kendilerinin, ürünlerde kinetik izotop etkilerine ve karşılık gelen izotopik zenginleşmelere sahip olan bazı diğer reaksiyonların ürünleri olması nedeniyle ortaya çıkar. Kinetik izotop etkisini belirlemek için NMR spektroskopisi kullanıldığında bu hatayı telafi etmek için aşağıdaki yönergeler önerilmiştir:[39][40]
- Referans olarak hizmet verecek ve reaksiyon merkezinden uzak bir karbon seçin ve reaksiyonda kinetik izotop etkisi olmadığını varsayın.
- Herhangi bir reaksiyona girmemiş başlangıç malzemesinde, diğer karbon NMR tepe integrallerinin referans karbonunkine oranlarını belirleyin.
- Bir miktar reaksiyona girdikten sonra başlangıç malzemesinin bir örneğindeki karbonlar için aynı oranları elde edin.
- İkinci oranların önceki oranlara oranları R / R verir0.
Jankowski tarafından listelenen diğer önlemlerin yanı sıra bunlara uyulursa, üç ondalık basamaklı hassasiyetlerle kinetik izotop etkileri elde edilebilir.[39][40]
Karbondan daha ağır elementlerle izotop etkileri
Karbon izotop etkilerinin yorumlanması, genellikle aynı anda karbona bağlar oluşturarak ve kırarak karmaşıklaşır. S gibi sadece karbondan bağ bölünmesini içeren reaksiyonlar bileN1 reaksiyon, kalan bağların karbonla güçlendirilmesini içerir. Bu tür birçok reaksiyonda, ayrılan grup izotop etkilerinin yorumlanması daha kolay olma eğilimindedir. Örneğin, klorun bir ayrılma grubu olarak hareket ettiği ikame ve eliminasyon reaksiyonlarının yorumlanması uygundur, çünkü klor, reaksiyon koordinatını karmaşıklaştırmak için hiçbir iç bağa sahip olmayan bir monatomik tür olarak işlev görür ve iki kararlı izotopu vardır, 35Cl ve 37Cl, her ikisi de bol bol. Bu tür izotop etkilerinin yorumlanmasındaki en büyük zorluk, ayrılan grubun çözülmesidir.[8]
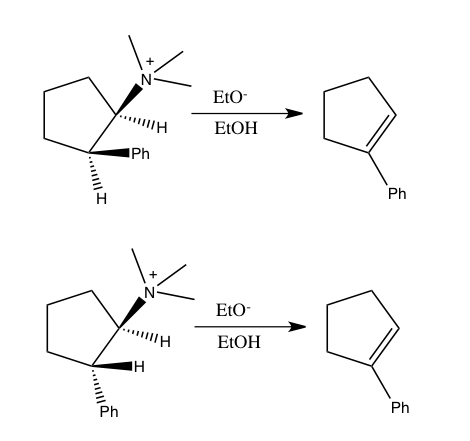
Deneysel belirsizlikler nedeniyle, izotop etkisinin ölçümü önemli belirsizliklere neden olabilir. Genellikle izotop etkileri, bir dizi izotopomer üzerinde tamamlayıcı çalışmalarla belirlenir. Buna göre, hidrojen izotop etkilerinin ağır atom izotop etkileriyle birleştirilmesi oldukça faydalıdır. Örneğin, nitrojen izotop etkisinin yanı sıra hidrojen izotop etkisinin belirlenmesi, 2-feniletiltrimetilamonyum iyonunun etanolde 40 ° C'de etoksit ile reaksiyonunun, alternatif uyumlu olmayan mekanizmaların aksine bir E2 mekanizmasını izlediğini göstermek için kullanıldı. Bu sonuca, bu reaksiyonun bir nitrojen izotop etkisi verdiğini göstererek ulaşıldı. k14/k151.0133 ± 0.0002, çıkan hidrojende 3.2 hidrojen kinetik izotop etkisi.[8]
Benzer şekilde, nitrojen ve hidrojen izotop etkilerinin birleştirilmesi, basit amonyum tuzlarının syn eliminasyonlarının da önceden tartışma konusu olan uyumlu bir mekanizmayı takip ettiğini göstermek için kullanıldı. 2-fenilsiklopentiltrimetilamonyum iyonunun etoksit ile aşağıdaki iki reaksiyonunda, her ikisi de 1-fenilsiklopenten verir, her iki izomer de bir nitrojen izotop etkisi gösterdi. k14/k15 60 ° C'de. Sin eliminasyonunu izleyen trans izomerin reaksiyonu, anti eliminasyona (1.0108) maruz kalan cis izomere kıyasla daha küçük bir nitrojen kinetik izotop etkisine (1.0064) sahip olsa da, her iki sonuç da CN bağının zayıfladığını gösterecek kadar büyüktür. uyumlu bir süreçte meydana gelebilecek geçiş durumunda.
Diğer örnekler
Kinetik izotop etkileri, izotopik kütlelerdeki farklılıklardan kaynaklandığından, en büyük gözlemlenebilir kinetik izotop etkileri, izotopik hidrojen ile döteryum (kütlede% 100 artış) veya trityum (kütlede% 200 artış) ile ilişkilidir. İzotopik kütle oranlarından kinetik izotop etkileri, müonlar kullanıldığında 36.4 kadar büyük olabilir. En hafif hidrojen atomunu ürettiler, 0.11H (0.113 amu), bir elektronun pozitif bir müon (μ+206 elektronluk bir kütleye sahip "çekirdek". Ayrıca, helyumdaki bir elektronu negatif bir müon (μ) ile değiştirerek en ağır hidrojen atomu analogunu hazırladılar.−) 4.116 amu'luk bir atom kütlesi ile Heu oluşturmak için. Negatif müon bir elektrondan çok daha ağır olduğu için, çekirdeğe çok daha yakın yörüngede dönerek bir protonu etkili bir şekilde korur ve Heu'nun şu şekilde davranmasını sağlar. 4.1H. Bu egzotik türlerle, H'nin 1H2 araştırılmıştır. En hafif ve en ağır hidrojen analoglarının reaksiyona girmesinden kaynaklanan sabit değerleri 1H2 daha sonra hesaplamak için kullanıldı k0.11/k4.1 izotopik kütlelerde 36.4 kat fark olduğu kinetik izotop etkisi. Bu reaksiyon için, izotopik ikame, ters kinetik izotop etkisi yaratır ve yazarlar, 1.74 x 10 kadar düşük bir kinetik izotop etkisi bildirirler.−4, şimdiye kadar bildirilen en küçük kinetik izotop etkisidir.[60]
Kinetik izotop etkisi, doğada sentezlendikleri yola bağlı olarak doğal ürünlerde döteryum izotoplarının belirli bir dağılımına yol açar. NMR spektroskopisi ile, şaraptaki alkolün fermente edilip edilmediğini tespit etmek kolaydır. glikoz veya yasadışı eklenmiş sakaroz.
Bir diğeri reaksiyon mekanizmaları kinetik izotop etkisi kullanılarak aydınlatılan, halojenleşme nın-nin toluen:[61]
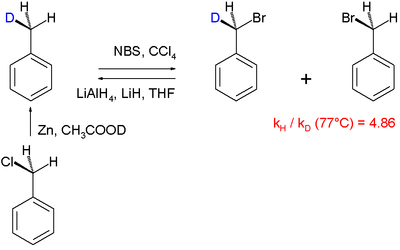
Bu özel "intramoleküler KIE" çalışmasında, bir benzilik hidrojen, radikal ikame brom kullanarak N-bromosüksinimid bromlama ajanı olarak. PhCH olduğu bulundu3 PhCD'den 4,86 kat daha hızlı brom yapar3. 5.56 değerinde büyük bir KIE, ketonlar ile brom ve sodyum hidroksit.[62]

Bu reaksiyonda hız sınırlayıcı aşama, enolate ketonun protonsuzlaştırılmasıyla. Bu çalışmada KIE, reaksiyon hızı sabitleri normal 2,4-dimetil-3-pentanon ve döteryumlanmış izomeri için optik yoğunluk ölçümler.
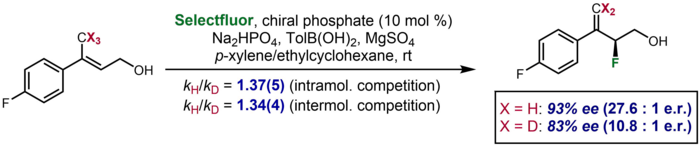
Asimetrik katalizde, bir kinetik izotop etkisinin döteryumlanmış olmayan bir alt tabakaya kıyasla döteryumlanmış bir alt tabaka için gözlemlenen enantioseçicilikte önemli bir fark olarak ortaya çıktığı ender durumlar vardır. Toste ve arkadaşları tarafından, döteryumlanmış bir substratın, döteryumlanmamış substrat için% 93 ee'ye kıyasla% 83 ee'lik bir enantioseçicilik ürettiği bir örnek bildirildi. Etki, enantiyodetme adımında C-H / D bağının bölünmesini öneren ek moleküller arası ve intramoleküler rekabet KIE verilerini desteklemek için alındı.[63]
Ayrıca bakınız
- Crossover deneyi (kimya)
- Denge sabiti # İzotopik ikame etkisi
- Manyetik izotop etkisi
- Reaksiyon mekanizması
- Geçici kinetik izotop fraksiyonasyonu
Referanslar
- ^ a b c d e Westaway KC (2006). "S'nin geçiş durumlarının yapısını belirlemek için kinetik izotop efektlerinin kullanılmasıN2 reaksiyon ". Fiziksel Organik Kimyadaki Gelişmeler. 41: 217–273. doi:10.1016 / S0065-3160 (06) 41004-2.
- ^ Lynn KR, Yankwich PE (5 Ağustos 1961). "Siyanür İyonunun Metil Klorür ve Metil Bromür ile Reaksiyonlarında Metil Karbonda İzotop Fraksiyonasyonu". Amerikan Kimya Derneği Dergisi. 83 (15): 3220–3223. doi:10.1021 / ja01476a012.
- ^ a b c d e f Atkins P, de Paula J (2006). Atkins'in Fiziksel Kimyası (8. baskı). Oxford University Press. pp.286 –288, 816–818. ISBN 978-0-19-870072-2.
- ^ a b c d e f g h Laidler KJ (1987). Kimyasal kinetik (3. baskı). Harper & Row. ISBN 978-0-06-043862-3.
- ^ a b c d Simmons EM, Hartwig JF (Mart 2012). "C-H Bağ İşlevselleştirmelerinde Döteryum Kinetik İzotop Etkilerinin Geçiş-Metal Kompleksleriyle Yorumlanması Üzerine". Angewandte Chemie Uluslararası Sürümü. 51 (1): 3066–72. doi:10.1002 / anie.201107334. PMID 22392731.
- ^ Poirier RA, Wang Y, Westaway KC (Mart 1994). "İkincil .alfa.-Döteryum Kinetik İzotop Etkileri ve S'nin Yapısı Arasındaki İlişkinin Teorik Bir Çalışması"N2 Geçiş Durumları ". Amerikan Kimya Derneği Dergisi. 116 (6): 2526–2533. doi:10.1021 / ja00085a037.
- ^ a b c d e Buncel E, Lee CC (1977). Katyonik reaksiyonlarda izotoplar. Organik Kimyada İzotoplar. 5. Amsterdam: Elsevier. ISBN 978-0-444-41927-9. OCLC 867217247.
- ^ a b c d e f g h ben Melander L, Saunders WH (1980). İzotopik Moleküllerin Reaksiyon Hızları. New York: Wiley.
- ^ Bigeleisen J, Wolfsberg M (Ocak 1957). Kimyasal kinetikte izotop etkilerinin "teorik ve deneysel yönleri". Kimyasal Fizikteki Gelişmeler. 1: 15–76.
- ^ Eğer müonyum (μ+e–) bir hidrojen izotopu olarak kabul edilir, bu durumda prensip olarak daha da büyük KIE'ler mümkündür. Bununla birlikte, muonyum içeren çalışmalar, müonun kısa yarı ömrü (22 mikrosaniye) ile sınırlıdır (bkz. Villà J, Corchado JC, González-Lafont A, Lluch JM, Truhlar DG (Kasım 1998). "Bir olefine hidrojen atomu ilavesi için döteryum ve muonyum kinetik izotop etkilerinin açıklaması". Amerikan Kimya Derneği Dergisi. 120 (46): 12141–2. doi:10.1021 / ja982616i. bir örnek için kMu/kH izotop etkisi.)
- ^ Bu kongre, hem isimlendirmede kolaylık hem de döteryum kinetik izotop etkilerinin deneysel olarak nasıl çalışıldığının bir yansıması olarak mevcuttur: Döteryum, IUPAC onaylı sembolü D = 2H, özellikle protium'u ifade eden ortak bir sembol yoktur (1H). Bununla birlikte, protium veya döteryum içeren izotopologların ilgili hız sabitlerine atıfta bulunmak için etiketlere sahip olmanın yararlı olduğu kanıtlanmıştır. kH ve kDsırasıyla tipik olarak kullanılmıştır. Dahası, kinetik izotop etkisinin büyüklüğü şu şekilde ifade edilebilir: kH/kD. Bu gösterim, deneysel olarak döteryum kinetik izotop etkilerinin, döteryumla zenginleştirilmiş bir başlangıç malzemesinin reaksiyon hızının, doğal bollukta hidrojen içeren zenginleştirilmemiş bir başlangıç malzemesinin reaksiyon hızıyla karşılaştırılmasıyla ölçüldüğü gerçeğiyle tutarlıdır. Protium doğal hidrojenin% 99.9885'ini oluşturduğundan, bu neredeyse her zaman geçerlidir, bu nedenle "protiumla zenginleştirilmiş" bir numune elde etmek için genellikle başlangıç malzemesindeki döteryumu daha fazla tüketmeye gerek yoktur. Birleştirildiğinde, gösterim ve deney düzeneği, döteryumun bir izotop etki çalışmasında "normal" hidrojenin yerini alan bir "ikame" olarak ortak kavramsallaştırılmasına yol açtı.
- ^ Bigeleisen J (Ağustos 1949). "İzotopik Moleküllerin Bağıl Tepkime Hızları". Kimyasal Fizik Dergisi. 17 (8): 675–678. Bibcode:1949JChPh..17..675B. doi:10.1063/1.1747368.
- ^ a b c Lowry TH, Richardson KS (1987). Organik kimyada mekanizma ve teori (3. baskı). New York: Harper & Row. pp.256. ISBN 978-0-06-044084-8. OCLC 14214254.
- ^ Carpenter BK (1984). Organik reaksiyon mekanizmalarının belirlenmesi. New York: Wiley. s. 86. ISBN 978-0-471-89369-1. OCLC 9894996.
- ^ Carpenter BK (Şubat 2010). "Kinetik izotop etkileri: sıradışı olanı ortaya çıkarmak". Doğa Kimyası. 2 (2): 80–2. Bibcode:2010 NatCh ... 2 ... 80C. doi:10.1038 / nchem.531. PMID 21124393.
- ^ Carroll FA (2010). Organik kimyada yapı ve mekanizma üzerine bakış açıları (2. baskı). Hoboken, NJ: John Wiley. ISBN 978-0-470-27610-5. OCLC 286483846.
- ^ Kwart H (1 Aralık 1982). "Mekanik bir kriter olarak birincil kinetik hidrojen izotop etkisinin sıcaklık bağımlılığı". Kimyasal Araştırma Hesapları. 15 (12): 401–408. doi:10.1021 / ar00084a004. ISSN 0001-4842.
- ^ Streitwieser A, Jagow RH, Fahey RC, Suzuki S (Mayıs 1958). "Döteryumlanmış siklopentil tosilatların asetolizlerinde kinetik izotop etkileri 1, 2". Amerikan Kimya Derneği Dergisi. 80 (9): 2326–32. doi:10.1021 / ja01542a075.
- ^ Swain CG, Stivers EC, Reuwer Jr JF, Schaad LJ (1 Kasım 1958). "Asetik Asit Tarafından Katalize Edilmiş Ketonların Enolizasyonunda Saldıran Nükleofili Tanımlamak için Hidrojen İzotop Etkilerinin Kullanımı". Amerikan Kimya Derneği Dergisi. 80 (21): 5885–5893. doi:10.1021 / ja01554a077.
- ^ a b c d e f g h ben j Anslyn EV, Dougherty DA (2006). Modern Fiziksel Organik Kimya. Üniversite Bilim Kitapları. pp.428 –437. ISBN 978-1-891389-31-3.
- ^ Razauy M (2003). Kuantum Tünel Teorisi. Dünya Bilimsel. ISBN 978-981-238-019-7.
- ^ Silbey RJ, Alberty RA, Bawendi MG (2005). Fiziksel kimya. John Wiley & Sons. s. 326–338. ISBN 978-0-471-21504-2.
- ^ Borgis D, Hynes JT (1993). "Çözümde proton tünelleme transfer hızlarının dinamik teorisi: Genel formülasyon". Kimyasal Fizik. 170 (3): 315–346. Bibcode:1993CP .... 170..315B. doi:10.1016 / 0301-0104 (93) 85117-Q.
- ^ a b Krishtalik LI (Mayıs 2000). "Proton transferinin mekanizması: bir taslak". Biochimica et Biophysica Açta. 1458 (1): 6–27. doi:10.1016 / S0005-2728 (00) 00057-8. PMID 10812022.
- ^ Zuev PS, Sheridan RS, Albu TV, Truhlar DG, Hrovat DA, Borden WT (Şubat 2003). "Tek bir kuantum durumundan karbon tünellemesi". Bilim. 299 (5608): 867–70. Bibcode:2003Sci ... 299..867Z. doi:10.1126 / science.1079294. PMID 12574623.
- ^ Fujisaki N, Ruf A, Gaeumann T (1987). "Hidrojen döteryum kinetik izotop etkilerinin sıcaklığa bağımlılığı ile incelenen hidrojen-atom-transfer reaksiyonlarında tünel etkileri". Journal of Physical Chemistry. 91 (6): 1602–1606. doi:10.1021 / j100290a062.
- ^ Lewis ES, Funderburk L (1967). "2-nitropropandan piridin bazlarına proton transferlerinde hızlar ve izotop etkileri". Amerikan Kimya Derneği Dergisi. 89 (10): 2322–2327. doi:10.1021 / ja00986a013.
- ^ Dewar MJ, Healy EF, Ruiz JM (1988). "1,3-pentadiende 1,5-sigmatropik hidrojen kaymasının mekanizması". Amerikan Kimya Derneği Dergisi. 110 (8): 2666–2667. doi:10.1021 / ja00216a060.
- ^ von Doering W, Zhao X (Temmuz 2006). "Cisoid kilitli 1,3 (Z) -pentadien, 2-metil-10-metilenbisiklo [4.4.0] dec-1-en'in 1,5-hidrojen kaymasında döteryumun kinetik üzerindeki etkisi: tünelleme için kanıt? ". Amerikan Kimya Derneği Dergisi. 128 (28): 9080–5. doi:10.1021 / ja057377v. PMID 16834382.
- ^ Bu çalışmada KIE, hassas proton NMR. 25 ° C'de tahmini KIE 16.6'dır, ancak hata payı yüksektir
- ^ Kohen A, Klinman JP (Temmuz 1999). "Biyolojide hidrojen tüneli". Kimya ve Biyoloji. 6 (7): R191-8. doi:10.1016 / S1074-5521 (99) 80058-1. PMID 10381408.
- ^ Wilde TC, Blotny G, Pollack RM (Mayıs 2008). "Ketosteroid izomeraz tarafından enzimle güçlendirilmiş bağlı hareket / kuantum mekanik hidrojen tüneli için deneysel kanıt". Amerikan Kimya Derneği Dergisi. 130 (20): 6577–85. doi:10.1021 / ja0732330. PMID 18426205.
- ^ Truhlar DG, Gao J, Alhambra C, Garcia-Viloca M, Corchado J, Sánchez M, Villà J (2002). "Enzim Kinetiği Modellemesinde Kuantum Etkilerinin Dahil Edilmesi". Kimyasal Araştırma Hesapları. 35 (6): 341–349. doi:10.1021 / ar0100226.
- ^ Kohen, A; Klinman, J. P (1998). "Enzim Katalizi: Klasik Paradigmaların Ötesinde". Kimyasal Araştırma Hesapları. 31 (7): 397–404. doi:10.1021 / ar9701225.
- ^ Maggi F, Riley WJ (2010). "Biyokimyasal kinetikte izotopolog ve izotopomer türleşmesi ve fraksiyonlanmasının matematiksel tedavisi". Geochimica et Cosmochimica Açta. 74 (6): 1823. Bibcode:2010GeCoA..74.1823M. doi:10.1016 / j.gca.2009.12.021.
- ^ Newall, A. Raymond; Hayes, John; Bethell, Donald (1 Ocak 1974). "Alifatik diazo-bileşiklerin ayrışmasında ara maddeler. Bölüm XI. Difenilmetilenin çözelti içindeki aminler ile reaksiyonu üzerine mekanik çalışmalar". Kimya Derneği Dergisi, Perkin İşlemleri 2. 0 (11): 1307–1312. doi:10.1039 / P29740001307. ISSN 1364-5471.
- ^ Buncel E, Lee CC (1977). Organik Kimyada Karbon-13. Organik Kimyada İzotoplar. 3. Amsterdam: Elsevier. ISBN 978-0-444-41472-4. OCLC 606113159.
- ^ a b c d Singleton DA, Thomas AA (Eylül 1995). "Doğal Bollukta Çoklu Küçük Kinetik İzotop Etkilerinin Yüksek Kesinlikle Eşzamanlı Belirlenmesi". Amerikan Kimya Derneği Dergisi. 117 (36): 9357–9358. doi:10.1021 / ja00141a030.
- ^ a b c Jankowski S (Ocak 2009). "İzotop etki çalışmalarında NMR spektroskopisinin uygulanması". NMR Spektroskopisine İlişkin Yıllık Raporlar. 68: 149–191. doi:10.1016 / S0066-4103 (09) 06803-3. ISBN 9780123810410.
- ^ a b c Kwan E. "CHEM 106 Ders Notları - Ders 14 - Hesaplamalı Kimya" (PDF). Alındı 2 Kasım 2013.
- ^ Martin GJ, Martin ML (1984). "Yüksek alan kantitatif 2H NMR ile incelendiği üzere doğal bolluk seviyesinde döteryum etiketlemesi". Tetrahedron Mektupları. 22 (36): 3525–3528. doi:10.1016 / s0040-4039 (01) 81948-1.
- ^ a b Pascal Jr RA, Baum MW, Wagner CK, Rodgers LR (Eylül 1984). "Doğal bolluk döteryum NMR spektroskopisi ile organik reaksiyonlarda döteryum kinetik izotop etkilerinin ölçülmesi". Amerikan Kimya Derneği Dergisi. 106 (18): 5377–5378. doi:10.1021 / ja00330a071.
- ^ Gajewski JJ, Peterson KB, Kagel JR, Huang YJ (Aralık 1989). "İkincil döteryum kinetik izotop etkilerinden Diels-Alder reaksiyonundaki geçiş durumu yapı değişimi. Neredeyse simetrik dienlerin ve dienofillerin reaksiyonu neredeyse eşzamanlıdır". Amerikan Kimya Derneği Dergisi. 111 (25): 9078–9081. doi:10.1021 / ja00207a013.
- ^ a b Colletto C, Islam S, Juliá-Hernández F, Larrosa I (Şubat 2016). "Tiyofenlerin ve Benzo [b] tiyofenlerin Oda Sıcaklığında Doğrudan β-Arilasyonu ve Heck-Tipi Bir Yol için Kinetik Kanıt". Amerikan Kimya Derneği Dergisi. 138 (5): 1677–83. doi:10.1021 / jacs.5b12242. PMC 4774971. PMID 26788885.
- ^ a b Frost GB, Serratore NA, Ogilvie JM, Douglas CJ (Nisan 2017). "Enantioselektif İntramoleküler Alken Siyanoamidasyonu için Palladyum Katalizeli C-CN Bağ Aktivasyonu için Mekanistik Model". Organik Kimya Dergisi. 82 (7): 3721–3726. doi:10.1021 / acs.joc.7b00196. PMC 5535300. PMID 28294618.
- ^ Kwan EE, Park Y, Besser HA, Anderson TL, Jacobsen EN (Ocak 2017). "Polarizasyon Transferiyle Etkinleştirilen 13C Kinetik İzotop Etkisi Ölçümleri". Amerikan Kimya Derneği Dergisi. 139 (1): 43–46. doi:10.1021 / jacs.6b10621. PMC 5674980. PMID 28005341.
- ^ Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN (Ocak 2017). "Makrosiklik bis-tiyoüreler stereospesifik glikosilasyon reaksiyonlarını katalize eder". Bilim. 355 (6321): 162–166. Bibcode:2017Sci ... 355..162P. doi:10.1126 / science.aal1875. PMC 5671764. PMID 28082586.
- ^ Burlingham BT, Pratt LM, Davidson ER, Shiner VJ, Fong J, Widlanski TS (Ekim 2003). "Sülfat ester hidrolizi üzerindeki 34S izotop etkisi: mekanik etkiler". Amerikan Kimya Derneği Dergisi. 125 (43): 13036–7. doi:10.1021 / ja0279747. PMID 14570471.
- ^ Hennig C, Oswald RB, Schmatz S (Mart 2006). "Nükleofilik ikamede ikincil kinetik izotop etkisi: kuantum mekanik bir yaklaşım". Fiziksel Kimya Dergisi A. 110 (9): 3071–9. Bibcode:2006JPCA..110.3071H. doi:10.1021 / jp0540151. PMID 16509628.
- ^ a b Cleland WW (Aralık 2003). "Enzim mekanizmalarını belirlemek için izotop etkilerinin kullanılması". Biyolojik Kimya Dergisi. 278 (52): 51975–84. doi:10.1074 / jbc.X300005200. PMID 14583616.
- ^ a b IUPAC, Kimyasal Terminoloji Özeti, 2. baskı. ("Altın Kitap") (1997). Çevrimiçi düzeltilmiş sürüm: (2006–) "İkincil izotop etkisi ". doi:10.1351 / goldbook.S05523
- ^ "İzotop etkisinin tanımı, ikincil". Kimya Sözlüğü. Chemitool.
- ^ Kurtz KA, Fitzpatrick PF (1997). "D-Amino Asit Oksidazın Nitroalkan Anyonlarla Reaksiyonu Üzerindeki pH ve İkincil Kinetik İzotop Etkileri: Karbanyonlar Tarafından Flavine Doğrudan Saldırının Kanıtı". Amerikan Kimya Derneği Dergisi. 119 (5): 1155–1156. doi:10.1021 / ja962783n.
- ^ Angelis YS, Hatzakis NS, Smonou I, Orfanopoulos M (2006). "Benzil alkollerin dimetildioksiran ile oksidasyonu. Kinetik izotop etkileriyle araştırılan uyumlu ve aşamalı mekanizmalar sorunu". Tetrahedron Mektupları. 42 (22): 3753–3756. doi:10.1016 / S0040-4039 (01) 00539-1.
- ^ a b Houk KN, Gustafson SM, Black KA (Ekim 1992). "Teorik ikincil kinetik izotop etkileri ve geçiş durumu geometrilerinin yorumlanması. 1. Cope yeniden düzenlenmesi". Amerikan Kimya Derneği Dergisi. 114 (22): 8565–72. doi:10.1021 / ja00048a032.
- ^ IUPAC, Kimyasal Terminoloji Özeti, 2. baskı. ("Altın Kitap") (1997). Çevrimiçi düzeltilmiş sürüm: (2006–) "sterik izotop etkisi ". doi:10.1351 / goldbook.S06001
- ^ Mislow K, Graeve R, Gordon AJ, Wahl GH (1963). "Sterik İzotop Etkileri Üzerine Bir Not. 9,10-Dihidro-4,5-Dimetilfenantren'in Rasemizasyonunda Konformasyonel Kinetik İzotop Etkileri". Amerikan Kimya Derneği Dergisi. 85 (8): 1199–1200. doi:10.1021 / ja00891a038.
- ^ Bartell LS (1 Eylül 1961). "İkincil İzotop Etkilerinde Bağlı Olmayan İtmelerin Rolü. I. Alfa ve Beta İkame Etkileri.1". Amerikan Kimya Derneği Dergisi. 83 (17): 3567–3571. doi:10.1021 / ja01478a006.
- ^ Felder T, Schalley CA (Mayıs 2003). "Rotaksanların dağılma reaksiyonu üzerindeki ikincil izotop etkileri: sterik boyutun yüksek hassasiyetli ölçümü". Angewandte Chemie. 42 (20): 2258–60. doi:10.1002 / anie.200350903. PMID 12772156.
- ^ Fleming DG, Arseneau DJ, Sukhorukov O, Brewer JH, Mielke SL, Schatz GC, Garrett BC, Peterson KA, Truhlar DG (Ocak 2011). "Muonik helyum ve muonyumun H2 ile reaksiyonları için kinetik izotop etkileri". Bilim. 331 (6016): 448–50. Bibcode:2011Sci ... 331..448F. doi:10.1126 / science.1199421. PMID 21273484.
- ^ Wiberg KB, Slaugh LH (1958). "Toluenin Yan Zincir Halojenasyonunda Döteryum İzotop Etkisi". Amerikan Kimya Derneği Dergisi. 80 (12): 3033–3039. doi:10.1021 / ja01545a034.
- ^ Lynch RA, Vincenti SP, Lin YT, Smucker LD, Subba Rao SC (1972). "Anormal kinetik hidrojen izotopunun, dialkil ikameli bazı ketonların iyonlaşma hızı üzerindeki etkileri". Amerikan Kimya Derneği Dergisi. 94 (24): 8351–8356. doi:10.1021 / ja00779a012.
- ^ Zi W, Wang YM, Toste FD (Eylül 2014). "Alilik alkollerin kiral anyon faz transfer florinasyonu için yerinde yönlendirici bir grup stratejisi". Amerikan Kimya Derneği Dergisi. 136 (37): 12864–7. doi:10.1021 / ja507468u. PMC 4183625. PMID 25203796.
daha fazla okuma
- Bell RP, Crooks JE (20 Temmuz 1965). "Bazı Ketonik Maddelerin İyonlaşmasında Kinetik Hidrojen İzotop Etkileri". Londra Kraliyet Cemiyeti Bildirileri A. 286 (1406): 285–299. Bibcode:1965RSPSA.286..285B. doi:10.1098 / rspa.1965.0144.